pMHC movement based on peptide anchoring
Introduction
In this notebook, apo-holo comparisons between different anchoring patterns of MHC alleles plotted next to each other. The anchoring patterns were determined by finding the peptide motifs of each MHC allele from the MHCMotifAtlas and using these strong motifs as anchors for the peptide.
[1]:
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import seaborn as sns
from python_pdb.formats.residue import THREE_TO_ONE_CODE
[ ]:
mhc_anchor_position_df = pd.read_csv('../data/external/mhc_motif_atlas.csv')
mhc_anchor_position_df = mhc_anchor_position_df.query("grade in ('high', 'dominant')")
mhc_anchor_position_df = mhc_anchor_position_df.sort_values('allele_slug')
mhc_anchor_position_df = mhc_anchor_position_df[['allele_slug', 'position', 'amino_acid', 'peptide_length']]
mhc_anchor_position_df = mhc_anchor_position_df.reset_index(drop=True)
mhc_anchor_position_df
| allele_slug | position | amino_acid | peptide_length | |
|---|---|---|---|---|
| 0 | hla_a_01_01 | 2 | T | 9 |
| 1 | hla_a_01_01 | 3 | D | 9 |
| 2 | hla_a_01_01 | 9 | Y | 9 |
| 3 | hla_a_02_01 | 2 | L | 9 |
| 4 | hla_a_02_01 | 9 | L | 9 |
| ... | ... | ... | ... | ... |
| 319 | hla_g_01_03 | 1 | K | 9 |
| 320 | hla_g_01_04 | 1 | R | 9 |
| 321 | hla_g_01_04 | 3 | P | 9 |
| 322 | hla_g_01_04 | 1 | K | 9 |
| 323 | hla_g_01_04 | 9 | L | 9 |
324 rows × 4 columns
Load apo-holo comparisons
[3]:
apo_holo_comparison = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I-comparisons/pmhc_per_res_apo_holo.csv')
peptide_apo_holo_comparison = apo_holo_comparison.query("chain_type == 'antigen_chain'").copy()
peptide_apo_holo_comparison
[3]:
| complex_id | structure_x_name | structure_y_name | chain_type | residue_name | residue_seq_id | residue_insert_code | rmsd | ca_distance | chi_angle_change | com_distance | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 181 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | MET | 1 | NaN | 0.700531 | 0.237162 | 1.570636 | 0.364628 |
| 182 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | VAL | 2 | NaN | 0.114569 | 0.089359 | 0.009407 | 0.043732 |
| 183 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | TRP | 3 | NaN | 0.400840 | 0.233532 | -0.019115 | 0.363989 |
| 184 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | GLY | 4 | NaN | 0.495618 | 0.262271 | NaN | 0.448252 |
| 185 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | PRO | 5 | NaN | 0.734430 | 0.486409 | -0.842595 | 0.536198 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 209372 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | ARG | 5 | NaN | 1.064663 | 0.157924 | 0.364432 | 0.705033 |
| 209373 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | THR | 6 | NaN | 0.421897 | 0.345439 | 0.097701 | 0.344074 |
| 209374 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | PHE | 7 | NaN | 1.225982 | 0.317819 | -0.347835 | 0.883236 |
| 209375 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | LEU | 8 | NaN | 1.323615 | 0.310356 | 0.612672 | 0.270049 |
| 209376 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | LEU | 9 | NaN | 1.599917 | 0.192969 | 0.885323 | 0.175807 |
9857 rows × 11 columns
[4]:
peptide_apo_holo_comparison['peptide_length'] = \
peptide_apo_holo_comparison.groupby(['complex_id',
'structure_x_name',
'structure_y_name']).transform('size')
[5]:
peptide_apo_holo_comparison['amino_acid'] = peptide_apo_holo_comparison['residue_name'].map(THREE_TO_ONE_CODE)
Load summary data
[6]:
summary_df = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I/apo_holo_summary.csv')
summary_df['group_name'] = summary_df['file_name'].str.replace('.pdb', '')
summary_df
/var/scratch/bmcmaste/2178229/ipykernel_2204500/1956598565.py:2: FutureWarning: The default value of regex will change from True to False in a future version.
summary_df['group_name'] = summary_df['file_name'].str.replace('.pdb', '')
[6]:
| file_name | pdb_id | structure_type | state | alpha_chain | beta_chain | antigen_chain | mhc_chain1 | mhc_chain2 | cdr_sequences_collated | peptide_sequence | mhc_slug | group_name | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 1ao7 | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVTTDSWGKLQ-MNHEY-SVGAGI-ASRPGLA... | LLFGYPVYV | hla_a_02_01 | 1ao7_D-E-C-A-B_tcr_pmhc |
| 1 | 1b0g_C-A-B_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | C | A | B | NaN | ALWGFFPVL | hla_a_02_01 | 1b0g_C-A-B_pmhc |
| 2 | 1b0g_F-D-E_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | F | D | E | NaN | ALWGFFPVL | hla_a_02_01 | 1b0g_F-D-E_pmhc |
| 3 | 1bd2_D-E-C-A-B_tcr_pmhc.pdb | 1bd2 | tcr_pmhc | holo | D | E | C | A | B | NSMFDY-ISSIKDK-AAMEGAQKLV-MNHEY-SVGAGI-ASSYPGG... | LLFGYPVYV | hla_a_02_01 | 1bd2_D-E-C-A-B_tcr_pmhc |
| 4 | 1bii_P-A-B_pmhc.pdb | 1bii | pmhc | apo | NaN | NaN | P | A | B | NaN | RGPGRAFVTI | h2_dd | 1bii_P-A-B_pmhc |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 386 | 7rtd_C-A-B_pmhc.pdb | 7rtd | pmhc | apo | NaN | NaN | C | A | B | NaN | YLQPRTFLL | hla_a_02_01 | 7rtd_C-A-B_pmhc |
| 387 | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 7rtr | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc |
| 388 | 8gvb_A-B-P-H-L_tcr_pmhc.pdb | 8gvb | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RYPLTFGW | hla_a_24_02 | 8gvb_A-B-P-H-L_tcr_pmhc |
| 389 | 8gvg_A-B-P-H-L_tcr_pmhc.pdb | 8gvg | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RFPLTFGW | hla_a_24_02 | 8gvg_A-B-P-H-L_tcr_pmhc |
| 390 | 8gvi_A-B-P-H-L_tcr_pmhc.pdb | 8gvi | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVVFTGGGNKLT-SEHNR-FQNEAQ-ASSL... | RYPLTFGW | hla_a_24_02 | 8gvi_A-B-P-H-L_tcr_pmhc |
391 rows × 13 columns
Annotate apo-holo data with allele information
[7]:
peptide_apo_holo_comparison = peptide_apo_holo_comparison.merge(summary_df, how='left', left_on='complex_id', right_on='group_name')
peptide_apo_holo_comparison
[7]:
| complex_id | structure_x_name | structure_y_name | chain_type | residue_name | residue_seq_id | residue_insert_code | rmsd | ca_distance | chi_angle_change | ... | state | alpha_chain | beta_chain | antigen_chain | mhc_chain1 | mhc_chain2 | cdr_sequences_collated | peptide_sequence | mhc_slug | group_name | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | MET | 1 | NaN | 0.700531 | 0.237162 | 1.570636 | ... | holo | D | E | C | A | B | NSAFQY-TYSSGN-AMRGDSSYKLI-SGHDY-FNNNVP-ASSLWEK... | MVWGPDPLYV | hla_a_02_01 | 5c0a_D-E-C-A-B_tcr_pmhc |
| 1 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | VAL | 2 | NaN | 0.114569 | 0.089359 | 0.009407 | ... | holo | D | E | C | A | B | NSAFQY-TYSSGN-AMRGDSSYKLI-SGHDY-FNNNVP-ASSLWEK... | MVWGPDPLYV | hla_a_02_01 | 5c0a_D-E-C-A-B_tcr_pmhc |
| 2 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | TRP | 3 | NaN | 0.400840 | 0.233532 | -0.019115 | ... | holo | D | E | C | A | B | NSAFQY-TYSSGN-AMRGDSSYKLI-SGHDY-FNNNVP-ASSLWEK... | MVWGPDPLYV | hla_a_02_01 | 5c0a_D-E-C-A-B_tcr_pmhc |
| 3 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | GLY | 4 | NaN | 0.495618 | 0.262271 | NaN | ... | holo | D | E | C | A | B | NSAFQY-TYSSGN-AMRGDSSYKLI-SGHDY-FNNNVP-ASSLWEK... | MVWGPDPLYV | hla_a_02_01 | 5c0a_D-E-C-A-B_tcr_pmhc |
| 4 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | PRO | 5 | NaN | 0.734430 | 0.486409 | -0.842595 | ... | holo | D | E | C | A | B | NSAFQY-TYSSGN-AMRGDSSYKLI-SGHDY-FNNNVP-ASSLWEK... | MVWGPDPLYV | hla_a_02_01 | 5c0a_D-E-C-A-B_tcr_pmhc |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 9852 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | ARG | 5 | NaN | 1.064663 | 0.157924 | 0.364432 | ... | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc |
| 9853 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | THR | 6 | NaN | 0.421897 | 0.345439 | 0.097701 | ... | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc |
| 9854 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | PHE | 7 | NaN | 1.225982 | 0.317819 | -0.347835 | ... | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc |
| 9855 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | LEU | 8 | NaN | 1.323615 | 0.310356 | 0.612672 | ... | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc |
| 9856 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | LEU | 9 | NaN | 1.599917 | 0.192969 | 0.885323 | ... | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc |
9857 rows × 26 columns
Combine anchoring information with apo-holo comparisons
[8]:
mhc_anchor_position_df['anchor'] = True
[9]:
peptide_apo_holo_comparison = peptide_apo_holo_comparison.merge(
mhc_anchor_position_df,
how='left',
left_on=['mhc_slug', 'residue_seq_id', 'amino_acid', 'peptide_length'],
right_on=['allele_slug', 'position', 'amino_acid', 'peptide_length'],
)
[10]:
def collate_anchors(group: pd.DataFrame) -> list[int]:
return sorted(group[group['anchor'] == True]['residue_seq_id'].unique().tolist())
anchoring_strategies = peptide_apo_holo_comparison.groupby(['structure_x_name', 'structure_y_name']).apply(collate_anchors)
anchoring_strategies.name = 'anchoring_strategy'
anchoring_strategies = anchoring_strategies.reset_index()
peptide_apo_holo_comparison = peptide_apo_holo_comparison.merge(anchoring_strategies, how='left', on=['structure_x_name', 'structure_y_name'])
[11]:
def markup_anchor(anchors: list[int]) -> str:
anchors = [f'p{anchor}' for anchor in anchors]
return '-'.join(anchors)
peptide_apo_holo_comparison['anchoring_strategy_str'] = peptide_apo_holo_comparison['anchoring_strategy'].map(markup_anchor)
[12]:
def find_dominant_anchor(group: pd.DataFrame) -> str:
anchor_types = group['anchoring_strategy_str'].unique()
lengths = np.array([len(anchor) for anchor in anchor_types])
index = np.argmax(lengths)
return anchor_types[index]
dominant_anchors = peptide_apo_holo_comparison.groupby('mhc_slug').apply(find_dominant_anchor)
dominant_anchors.name = 'dominant_anchor'
dominant_anchors = dominant_anchors.reset_index()
peptide_apo_holo_comparison = peptide_apo_holo_comparison.merge(dominant_anchors, how='left', on='mhc_slug')
The below correction is applied as it is assumed that those peptide marked as solely ‘p2’ or ‘p9’ anchors are most likely still anchored by something at either end that is not necessarily the high or dominant motif.
[13]:
peptide_apo_holo_comparison['dominant_anchor'] = peptide_apo_holo_comparison['dominant_anchor'].map(
lambda strategy: 'p2-p9' if strategy in ('p2', 'p9') else strategy
)
[14]:
peptide_apo_holo_comparison_with_anchor = peptide_apo_holo_comparison.query("anchoring_strategy_str != ''")
Visualising the results
[15]:
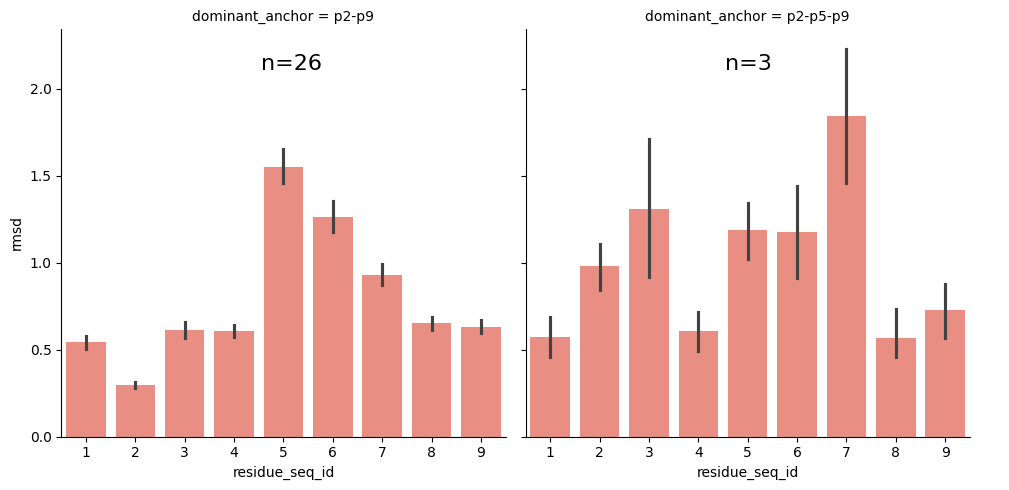
g = sns.catplot(peptide_apo_holo_comparison_with_anchor,
col='dominant_anchor',
x='residue_seq_id', y='rmsd',
sharex=False,
color='salmon',
kind='bar')
def annotate(data, **kws):
groups = data.groupby(['mhc_slug', 'peptide_sequence'])
ax = plt.gca()
ax.text(0.45, 0.9, f'n={len(groups)}', fontsize=16, transform=ax.transAxes)
g.map_dataframe(annotate)
[15]:
<seaborn.axisgrid.FacetGrid at 0x7f6443502380>
[16]:
max_rmsd = peptide_apo_holo_comparison_with_anchor['rmsd'].max()
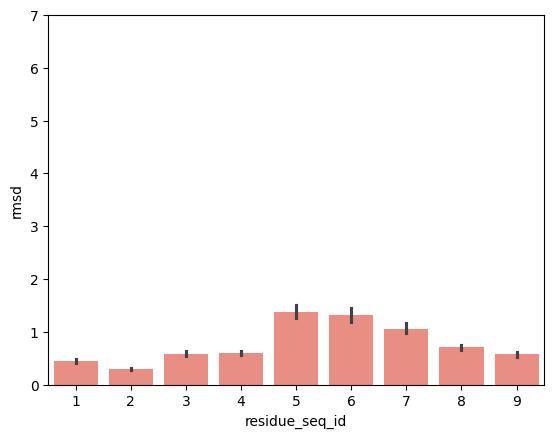
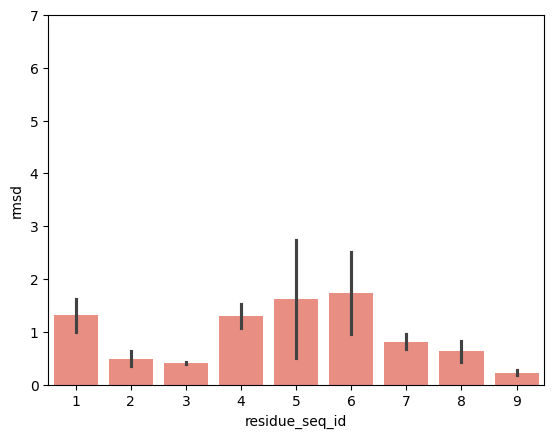
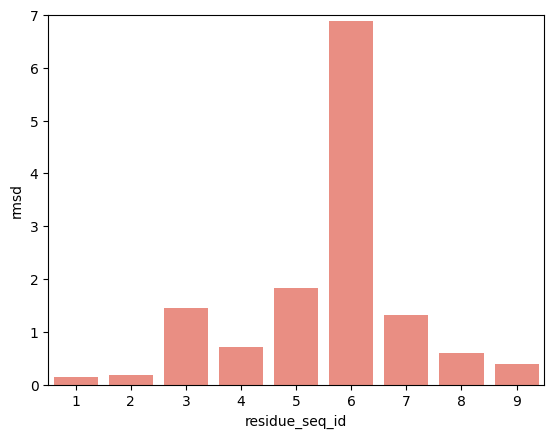
for mhc_slug, group in peptide_apo_holo_comparison_with_anchor.groupby('mhc_slug'):
print(mhc_slug)
print(group['dominant_anchor'].unique()[0])
plot = sns.barplot(group, x='residue_seq_id', y='rmsd', color='salmon')
plot.set_ylim((0, np.ceil(max_rmsd)))
plt.show()
hla_a_02_01
p2-p9
hla_a_24_02
p2-p9
hla_b_07_02
p2-p9
hla_b_08_01
p2-p5-p9
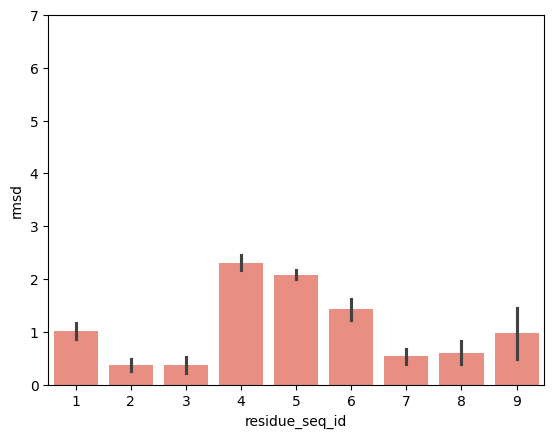
hla_b_35_01
p2-p9
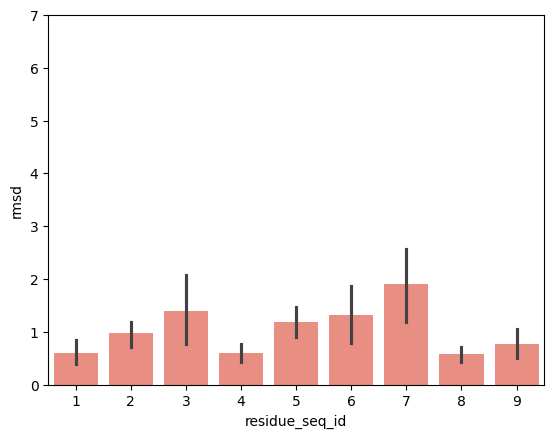
hla_b_37_01
p2-p5-p9
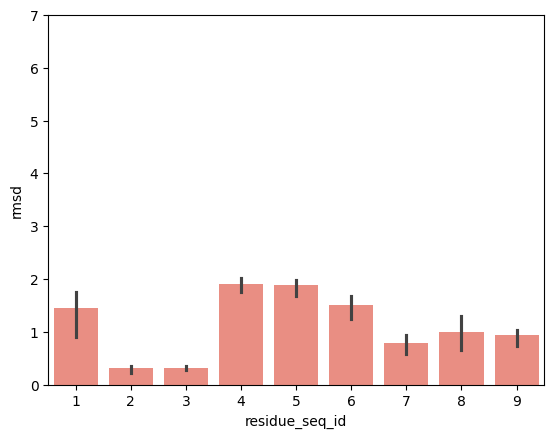
hla_b_42_01
p2-p9
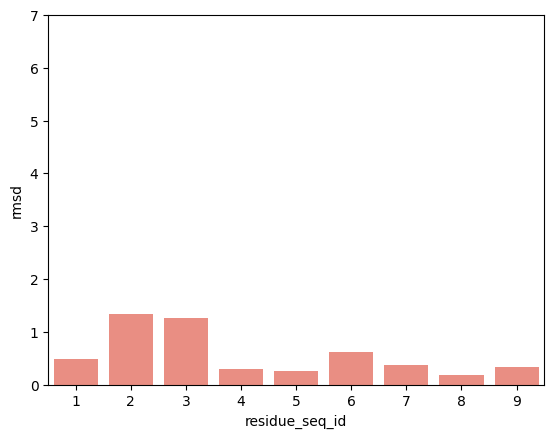
hla_b_44_05
p2-p9
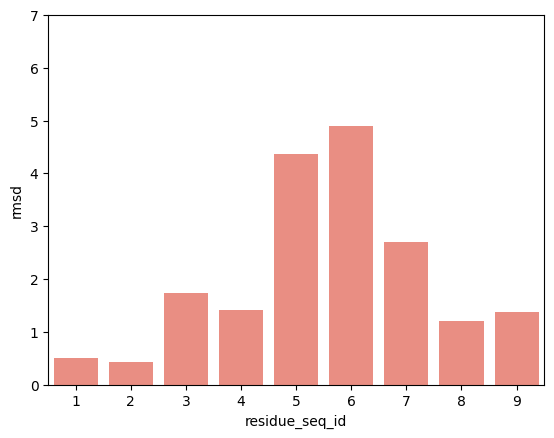
hla_b_53_01
p2-p9
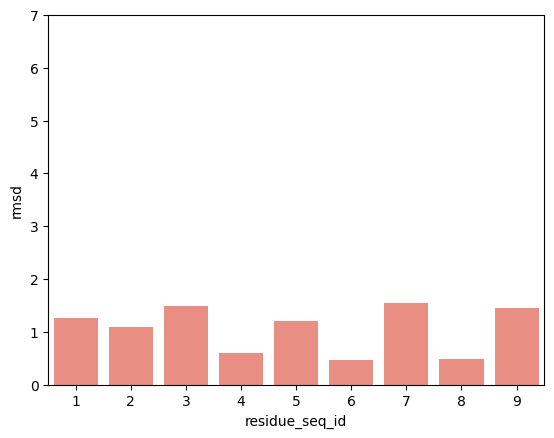
hla_b_81_01
p2-p9
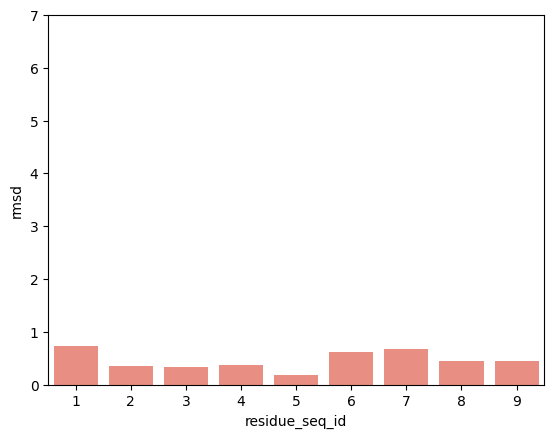
hla_e_01_03
p2-p9
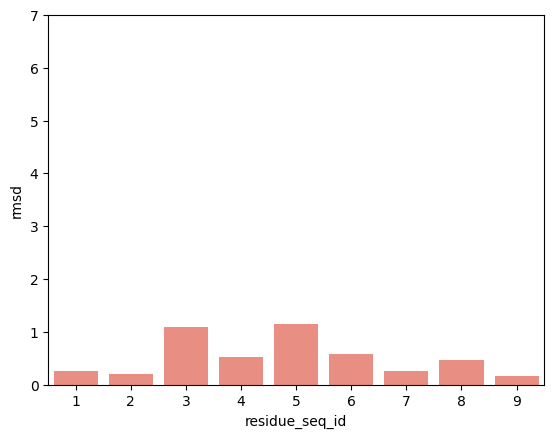
[17]:
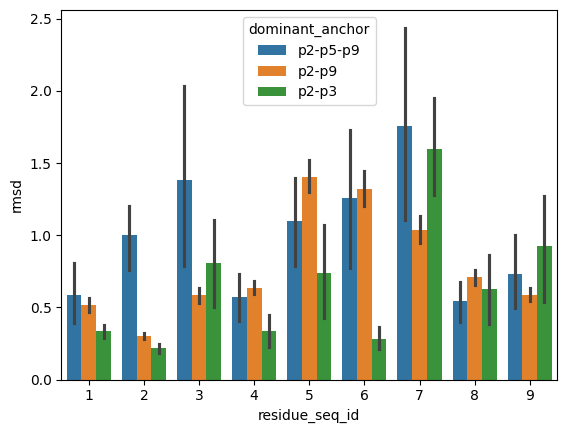
sns.barplot(peptide_apo_holo_comparison_with_anchor,
hue='dominant_anchor',
x='residue_seq_id', y='rmsd')
[17]:
<AxesSubplot: xlabel='residue_seq_id', ylabel='rmsd'>
[ ]:
representative_alleles = (peptide_apo_holo_comparison_with_anchor.groupby('dominant_anchor')[['dominant_anchor', 'mhc_slug']]
.sample(1, random_state=1)
.reset_index(drop=True))
representative_alleles
| dominant_anchor | mhc_slug | |
|---|---|---|
| 0 | p2-p5-p9 | hla_b_08_01 |
| 1 | p2-p9 | hla_e_01_03 |
[19]:
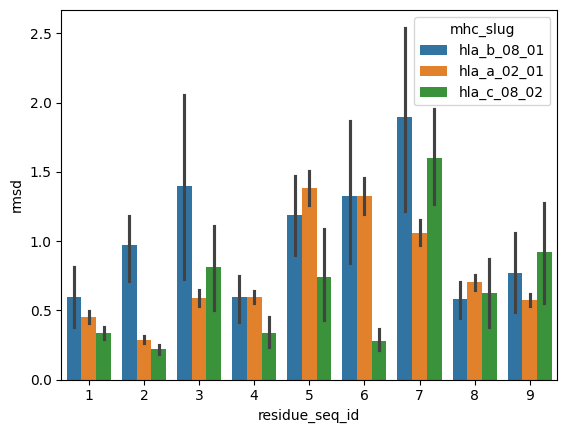
sns.barplot(peptide_apo_holo_comparison_with_anchor.query("mhc_slug.isin(@representative_alleles['mhc_slug'])"),
hue='mhc_slug',
x='residue_seq_id', y='rmsd')
[19]:
<AxesSubplot: xlabel='residue_seq_id', ylabel='rmsd'>
Conclusion
From the visualisations it is clear that the profile of peptide conformational changes depends on the mhc allele and how the peptides are anchored by the allele.