Length dependency of conformational change
Introduction
In this notebook, we set out to determine if there was a correlation between conformational change and the length of CDR loops or peptides. We looked at both the correlation of bulk movements when loops where aligned on the framework regions and also the deformation effects when loops are aligned to one another. All peptide conformations are measure from alignment on the antigen binding groove floor.
[1]:
import os
import matplotlib.pyplot as plt
import pandas as pd
import numpy as np
import scipy
import seaborn as sns
Loading Meta data
[2]:
DATA_DIR = '../data/processed/apo-holo-tcr-pmhc-class-I-comparisons'
[3]:
apo_holo_summary_df = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I/apo_holo_summary.csv')
apo_holo_summary_df['id'] = apo_holo_summary_df['file_name'].str.replace('.pdb$', '', regex=True)
[4]:
cdr_types = ['CDR-A1', 'CDR-A2', 'CDR-A3','CDR-B1', 'CDR-B2', 'CDR-B3']
apo_holo_summary_df[cdr_types] = apo_holo_summary_df['cdr_sequences_collated'].str.split('-').apply(pd.Series)
[5]:
apo_holo_summary_df
[5]:
| file_name | pdb_id | structure_type | state | alpha_chain | beta_chain | antigen_chain | mhc_chain1 | mhc_chain2 | cdr_sequences_collated | peptide_sequence | mhc_slug | id | CDR-A1 | CDR-A2 | CDR-A3 | CDR-B1 | CDR-B2 | CDR-B3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 1ao7 | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVTTDSWGKLQ-MNHEY-SVGAGI-ASRPGLA... | LLFGYPVYV | hla_a_02_01 | 1ao7_D-E-C-A-B_tcr_pmhc | DRGSQS | IYSNGD | AVTTDSWGKLQ | MNHEY | SVGAGI | ASRPGLAGGRPEQY |
| 1 | 1b0g_C-A-B_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | C | A | B | NaN | ALWGFFPVL | hla_a_02_01 | 1b0g_C-A-B_pmhc | NaN | NaN | NaN | NaN | NaN | NaN |
| 2 | 1b0g_F-D-E_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | F | D | E | NaN | ALWGFFPVL | hla_a_02_01 | 1b0g_F-D-E_pmhc | NaN | NaN | NaN | NaN | NaN | NaN |
| 3 | 1bd2_D-E-C-A-B_tcr_pmhc.pdb | 1bd2 | tcr_pmhc | holo | D | E | C | A | B | NSMFDY-ISSIKDK-AAMEGAQKLV-MNHEY-SVGAGI-ASSYPGG... | LLFGYPVYV | hla_a_02_01 | 1bd2_D-E-C-A-B_tcr_pmhc | NSMFDY | ISSIKDK | AAMEGAQKLV | MNHEY | SVGAGI | ASSYPGGGFYEQY |
| 4 | 1bii_P-A-B_pmhc.pdb | 1bii | pmhc | apo | NaN | NaN | P | A | B | NaN | RGPGRAFVTI | h2_dd | 1bii_P-A-B_pmhc | NaN | NaN | NaN | NaN | NaN | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 386 | 7rtd_C-A-B_pmhc.pdb | 7rtd | pmhc | apo | NaN | NaN | C | A | B | NaN | YLQPRTFLL | hla_a_02_01 | 7rtd_C-A-B_pmhc | NaN | NaN | NaN | NaN | NaN | NaN |
| 387 | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 7rtr | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc | DRGSQS | IYSNGD | AVNRDDKII | SEHNR | FQNEAQ | ASSPDIEQY |
| 388 | 8gvb_A-B-P-H-L_tcr_pmhc.pdb | 8gvb | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RYPLTFGW | hla_a_24_02 | 8gvb_A-B-P-H-L_tcr_pmhc | YGATPY | YFSGDTLV | AVGFTGGGNKLT | SEHNR | FQNEAQ | ASSDRDRVPETQY |
| 389 | 8gvg_A-B-P-H-L_tcr_pmhc.pdb | 8gvg | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RFPLTFGW | hla_a_24_02 | 8gvg_A-B-P-H-L_tcr_pmhc | YGATPY | YFSGDTLV | AVGFTGGGNKLT | SEHNR | FQNEAQ | ASSDRDRVPETQY |
| 390 | 8gvi_A-B-P-H-L_tcr_pmhc.pdb | 8gvi | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVVFTGGGNKLT-SEHNR-FQNEAQ-ASSL... | RYPLTFGW | hla_a_24_02 | 8gvi_A-B-P-H-L_tcr_pmhc | YGATPY | YFSGDTLV | AVVFTGGGNKLT | SEHNR | FQNEAQ | ASSLRDRVPETQY |
391 rows × 19 columns
TCR Analysis
Load Data
[6]:
results_tcr_fw_align = pd.read_csv(os.path.join(DATA_DIR, 'rmsd_cdr_fw_align_results.csv'))
results_tcr_fw_align['alignment'] = 'framework'
[7]:
results_tcr_loop_align = pd.read_csv(os.path.join(DATA_DIR, 'rmsd_cdr_loop_align_results.csv'))
results_tcr_loop_align['alignment'] = 'loop'
[8]:
results_tcr = pd.concat([results_tcr_fw_align, results_tcr_loop_align])
results_tcr
[8]:
| complex_id | structure_x_name | structure_y_name | chain_type | cdr | rmsd | alignment | |
|---|---|---|---|---|---|---|---|
| 0 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | 1.932806 | framework |
| 1 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 2 | 1.308598 | framework |
| 2 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 3 | 1.244062 | framework |
| 3 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | beta_chain | 1 | 0.809066 | framework |
| 4 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | beta_chain | 2 | 0.688597 | framework |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 809 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | alpha_chain | 2 | 0.206414 | loop |
| 810 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | alpha_chain | 3 | 0.459090 | loop |
| 811 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 1 | 0.255668 | loop |
| 812 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 2 | 0.175123 | loop |
| 813 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | 0.223801 | loop |
1628 rows × 7 columns
Merge with metadata
[9]:
apo_holo_summary_cdrs_df = apo_holo_summary_df.melt(
id_vars=[col for col in apo_holo_summary_df.columns if col not in cdr_types],
var_name='cdr_type',
value_name='cdr_sequence',
value_vars=cdr_types,
)
[10]:
apo_holo_summary_cdrs_df[['chain_type', 'cdr']] = apo_holo_summary_cdrs_df['cdr_type'].map(
lambda cdr_type: ('alpha_chain' if cdr_type[-2] == 'A' else 'beta_chain', int(cdr_type[-1]))
).apply(pd.Series)
[11]:
apo_holo_summary_cdrs_df['cdr_length'] = apo_holo_summary_cdrs_df['cdr_sequence'].str.len()
[12]:
apo_holo_summary_cdrs_df
[12]:
| file_name | pdb_id | structure_type | state | alpha_chain | beta_chain | antigen_chain | mhc_chain1 | mhc_chain2 | cdr_sequences_collated | peptide_sequence | mhc_slug | id | cdr_type | cdr_sequence | chain_type | cdr | cdr_length | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 1ao7 | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVTTDSWGKLQ-MNHEY-SVGAGI-ASRPGLA... | LLFGYPVYV | hla_a_02_01 | 1ao7_D-E-C-A-B_tcr_pmhc | CDR-A1 | DRGSQS | alpha_chain | 1 | 6.0 |
| 1 | 1b0g_C-A-B_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | C | A | B | NaN | ALWGFFPVL | hla_a_02_01 | 1b0g_C-A-B_pmhc | CDR-A1 | NaN | alpha_chain | 1 | NaN |
| 2 | 1b0g_F-D-E_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | F | D | E | NaN | ALWGFFPVL | hla_a_02_01 | 1b0g_F-D-E_pmhc | CDR-A1 | NaN | alpha_chain | 1 | NaN |
| 3 | 1bd2_D-E-C-A-B_tcr_pmhc.pdb | 1bd2 | tcr_pmhc | holo | D | E | C | A | B | NSMFDY-ISSIKDK-AAMEGAQKLV-MNHEY-SVGAGI-ASSYPGG... | LLFGYPVYV | hla_a_02_01 | 1bd2_D-E-C-A-B_tcr_pmhc | CDR-A1 | NSMFDY | alpha_chain | 1 | 6.0 |
| 4 | 1bii_P-A-B_pmhc.pdb | 1bii | pmhc | apo | NaN | NaN | P | A | B | NaN | RGPGRAFVTI | h2_dd | 1bii_P-A-B_pmhc | CDR-A1 | NaN | alpha_chain | 1 | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 2341 | 7rtd_C-A-B_pmhc.pdb | 7rtd | pmhc | apo | NaN | NaN | C | A | B | NaN | YLQPRTFLL | hla_a_02_01 | 7rtd_C-A-B_pmhc | CDR-B3 | NaN | beta_chain | 3 | NaN |
| 2342 | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 7rtr | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | 7rtr_D-E-C-A-B_tcr_pmhc | CDR-B3 | ASSPDIEQY | beta_chain | 3 | 9.0 |
| 2343 | 8gvb_A-B-P-H-L_tcr_pmhc.pdb | 8gvb | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RYPLTFGW | hla_a_24_02 | 8gvb_A-B-P-H-L_tcr_pmhc | CDR-B3 | ASSDRDRVPETQY | beta_chain | 3 | 13.0 |
| 2344 | 8gvg_A-B-P-H-L_tcr_pmhc.pdb | 8gvg | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RFPLTFGW | hla_a_24_02 | 8gvg_A-B-P-H-L_tcr_pmhc | CDR-B3 | ASSDRDRVPETQY | beta_chain | 3 | 13.0 |
| 2345 | 8gvi_A-B-P-H-L_tcr_pmhc.pdb | 8gvi | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVVFTGGGNKLT-SEHNR-FQNEAQ-ASSL... | RYPLTFGW | hla_a_24_02 | 8gvi_A-B-P-H-L_tcr_pmhc | CDR-B3 | ASSLRDRVPETQY | beta_chain | 3 | 13.0 |
2346 rows × 18 columns
[13]:
results_tcr = results_tcr.merge(
apo_holo_summary_cdrs_df[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary_cdrs_df[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
).merge(
apo_holo_summary_cdrs_df[['id',
'cdr_sequences_collated',
'peptide_sequence',
'mhc_slug',
'cdr_sequence',
'cdr_length',
'cdr_type',
'chain_type',
'cdr']],
how='left',
left_on=['complex_id', 'chain_type', 'cdr'],
right_on=['id', 'chain_type', 'cdr'],
)
Normalise data
[14]:
results_tcr['comparison'] = results_tcr['state_x'] + '-' + results_tcr['state_y']
results_tcr['comparison'] = results_tcr['comparison'].map(lambda entry: 'apo-holo' if entry == 'holo-apo' else entry)
results_tcr = results_tcr.query("comparison == 'apo-holo'").reset_index(drop=True)
[15]:
results_tcr['structure_comparison'] = results_tcr.apply(
lambda row: '-'.join(sorted([row.structure_x_name, row.structure_y_name])),
axis='columns',
)
results_tcr = results_tcr.drop_duplicates(['structure_comparison', 'chain_type', 'cdr', 'alignment'])
[16]:
results_tcr = results_tcr.groupby(['cdr_sequences_collated',
'alignment',
'comparison',
'cdr_length',
'cdr_sequence',
'cdr_type',
'chain_type',
'cdr'])['rmsd'].mean().reset_index()
Visualise Results
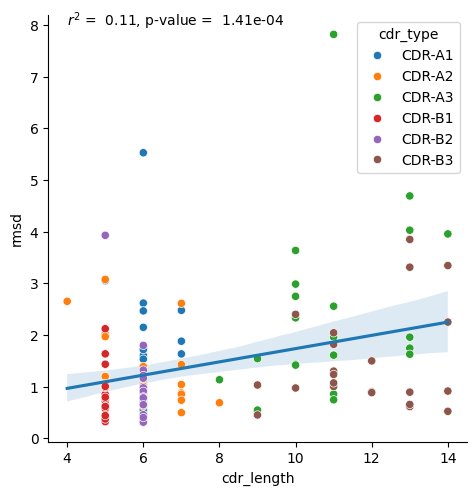
Alignment on Framework region
[17]:
data = results_tcr.query("alignment == 'framework'")
r, p_val = scipy.stats.pearsonr(data['cdr_length'], data['rmsd'])
sns.lmplot(data.sort_values('cdr_type'), x='cdr_length', y='rmsd', scatter=False)
sns.scatterplot(data.sort_values('cdr_type'), x='cdr_length', y='rmsd', hue='cdr_type')
plt.text(4, 8, f'$r^2$ = {r**2: .2f}, p-value = {p_val: .2e}')
plt.show()
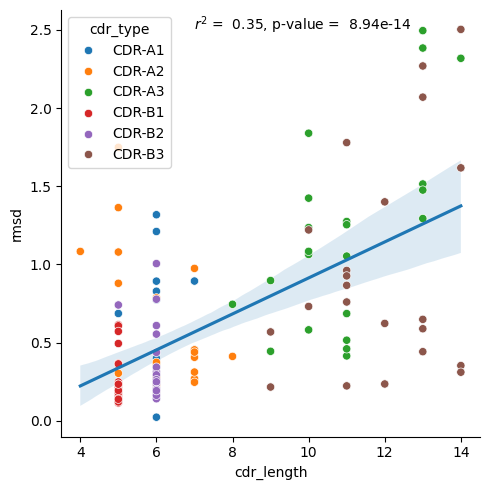
Alignment on loops
[18]:
data = results_tcr.query("alignment == 'loop'")
r, p_val = scipy.stats.pearsonr(data['cdr_length'], data['rmsd'])
sns.lmplot(data.sort_values('cdr_type'), x='cdr_length', y='rmsd', scatter=False)
sns.scatterplot(data.sort_values('cdr_type'), x='cdr_length', y='rmsd', hue='cdr_type')
plt.text(7, 2.5, f'$r^2$ = {r**2: .2f}, p-value = {p_val: .2e}')
plt.show()
TCR apex calculations
[19]:
results_per_res_tcr = pd.read_csv(os.path.join(DATA_DIR, 'tcr_per_res_apo_holo_loop_align.csv'))
results_per_res_tcr
[19]:
| complex_id | structure_x_name | structure_y_name | chain_type | cdr | residue_name | residue_seq_id | residue_insert_code | rmsd | ca_distance | chi_angle_change | com_distance | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | ASP | 27 | NaN | 4.922807 | 2.215234 | -1.001709 | 3.836500 |
| 1 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | ARG | 28 | NaN | 7.683418 | 2.322292 | -1.010462 | 6.119157 |
| 2 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | GLY | 29 | NaN | 0.657793 | 0.718576 | NaN | 0.452200 |
| 3 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | SER | 36 | NaN | 1.224430 | 0.404912 | -2.505061 | 0.866544 |
| 4 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | GLN | 37 | NaN | 1.133408 | 0.467132 | 0.667185 | 0.798590 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 6228 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | ASP | 109 | NaN | 0.507077 | 0.180564 | 0.198956 | 0.208659 |
| 6229 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | ILE | 114 | NaN | 2.164965 | 0.158682 | 3.679442 | 0.829175 |
| 6230 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | GLU | 115 | NaN | 1.577728 | 0.195887 | 3.091039 | 0.995351 |
| 6231 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | GLN | 116 | NaN | 0.204783 | 0.197683 | -0.014126 | 0.166472 |
| 6232 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | TYR | 117 | NaN | 0.180118 | 0.164981 | -0.024244 | 0.170852 |
6233 rows × 12 columns
[20]:
results_per_res_tcr = results_per_res_tcr.merge(
apo_holo_summary_cdrs_df[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary_cdrs_df[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
).merge(
apo_holo_summary_cdrs_df[['id',
'cdr_sequences_collated',
'peptide_sequence',
'mhc_slug',
'cdr_sequence',
'cdr_length',
'cdr_type',
'chain_type',
'cdr']],
how='left',
left_on=['complex_id', 'chain_type', 'cdr'],
right_on=['id', 'chain_type', 'cdr'],
)
[21]:
results_per_res_tcr['comparison'] = results_per_res_tcr['state_x'] + '-' + results_per_res_tcr['state_y']
results_per_res_tcr['comparison'] = results_per_res_tcr['comparison'].map(
lambda entry: 'apo-holo' if entry == 'holo-apo' else entry
)
results_per_res_tcr = results_per_res_tcr.query("comparison == 'apo-holo'").reset_index(drop=True)
[22]:
results_per_res_tcr['structure_comparison'] = results_per_res_tcr.apply(
lambda row: '-'.join(sorted([row.structure_x_name, row.structure_y_name])),
axis='columns',
)
results_per_res_tcr = results_per_res_tcr.drop_duplicates(['structure_comparison', 'chain_type', 'cdr',
'residue_name', 'residue_seq_id', 'residue_insert_code'])
[23]:
results_per_res_tcr
[23]:
| complex_id | structure_x_name | structure_y_name | chain_type | cdr | residue_name | residue_seq_id | residue_insert_code | rmsd | ca_distance | ... | state_y | id | cdr_sequences_collated | peptide_sequence | mhc_slug | cdr_sequence | cdr_length | cdr_type | comparison | structure_comparison | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | ASP | 27 | NaN | 4.922807 | 2.215234 | ... | apo | 3qdg_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNFGGGKLI-MRHNA-SNTAGT-ASSLSFGTEAF | ELAGIGILTV | hla_a_02_01 | DRGSQS | 6.0 | CDR-A1 | apo-holo | 3qdg_D-E-C-A-B_tcr_pmhc.pdb-3qeu_A-B_tcr.pdb |
| 36 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | ARG | 28 | NaN | 7.683418 | 2.322292 | ... | apo | 3qdg_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNFGGGKLI-MRHNA-SNTAGT-ASSLSFGTEAF | ELAGIGILTV | hla_a_02_01 | DRGSQS | 6.0 | CDR-A1 | apo-holo | 3qdg_D-E-C-A-B_tcr_pmhc.pdb-3qeu_A-B_tcr.pdb |
| 72 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | GLY | 29 | NaN | 0.657793 | 0.718576 | ... | apo | 3qdg_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNFGGGKLI-MRHNA-SNTAGT-ASSLSFGTEAF | ELAGIGILTV | hla_a_02_01 | DRGSQS | 6.0 | CDR-A1 | apo-holo | 3qdg_D-E-C-A-B_tcr_pmhc.pdb-3qeu_A-B_tcr.pdb |
| 108 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | SER | 36 | NaN | 1.224430 | 0.404912 | ... | apo | 3qdg_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNFGGGKLI-MRHNA-SNTAGT-ASSLSFGTEAF | ELAGIGILTV | hla_a_02_01 | DRGSQS | 6.0 | CDR-A1 | apo-holo | 3qdg_D-E-C-A-B_tcr_pmhc.pdb-3qeu_A-B_tcr.pdb |
| 144 | 3qdg_D-E-C-A-B_tcr_pmhc | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | 3qeu_A-B_tcr.pdb | alpha_chain | 1 | GLN | 37 | NaN | 1.133408 | 0.467132 | ... | apo | 3qdg_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNFGGGKLI-MRHNA-SNTAGT-ASSLSFGTEAF | ELAGIGILTV | hla_a_02_01 | DRGSQS | 6.0 | CDR-A1 | apo-holo | 3qdg_D-E-C-A-B_tcr_pmhc.pdb-3qeu_A-B_tcr.pdb |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 158220 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | ASP | 109 | NaN | 0.507077 | 0.180564 | ... | holo | 7rtr_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | ASSPDIEQY | 9.0 | CDR-B3 | apo-holo | 7n1d_A-B_tcr.pdb-7rtr_D-E-C-A-B_tcr_pmhc.pdb |
| 158256 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | ILE | 114 | NaN | 2.164965 | 0.158682 | ... | holo | 7rtr_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | ASSPDIEQY | 9.0 | CDR-B3 | apo-holo | 7n1d_A-B_tcr.pdb-7rtr_D-E-C-A-B_tcr_pmhc.pdb |
| 158292 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | GLU | 115 | NaN | 1.577728 | 0.195887 | ... | holo | 7rtr_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | ASSPDIEQY | 9.0 | CDR-B3 | apo-holo | 7n1d_A-B_tcr.pdb-7rtr_D-E-C-A-B_tcr_pmhc.pdb |
| 158328 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | GLN | 116 | NaN | 0.204783 | 0.197683 | ... | holo | 7rtr_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | ASSPDIEQY | 9.0 | CDR-B3 | apo-holo | 7n1d_A-B_tcr.pdb-7rtr_D-E-C-A-B_tcr_pmhc.pdb |
| 158364 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n1d_A-B_tcr.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | beta_chain | 3 | TYR | 117 | NaN | 0.180118 | 0.164981 | ... | holo | 7rtr_D-E-C-A-B_tcr_pmhc | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 | ASSPDIEQY | 9.0 | CDR-B3 | apo-holo | 7n1d_A-B_tcr.pdb-7rtr_D-E-C-A-B_tcr_pmhc.pdb |
4400 rows × 29 columns
[24]:
def compute_apex_movement(loop: pd.DataFrame) -> float:
loop_len = len(loop)
if loop_len % 2 == 0:
return np.mean([loop.iloc[loop_len // 2]['ca_distance'], loop.iloc[(loop_len // 2) - 1]['ca_distance']])
return loop.iloc[loop_len // 2]['ca_distance']
[25]:
results_tcr_apex = results_per_res_tcr.groupby(['structure_x_name',
'structure_y_name',
'cdr_type',
'cdr',
'chain_type',
'cdr_length']).apply(compute_apex_movement)
results_tcr_apex.name = 'apex_ca_distance'
results_tcr_apex = results_tcr_apex.reset_index()
[26]:
results_tcr_apex
[26]:
| structure_x_name | structure_y_name | cdr_type | cdr | chain_type | cdr_length | apex_ca_distance | |
|---|---|---|---|---|---|---|---|
| 0 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 3qh3_A-B_tcr.pdb | CDR-A1 | 1 | alpha_chain | 6.0 | 0.153049 |
| 1 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 3qh3_A-B_tcr.pdb | CDR-A2 | 2 | alpha_chain | 6.0 | 0.447122 |
| 2 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 3qh3_A-B_tcr.pdb | CDR-A3 | 3 | alpha_chain | 11.0 | 2.461146 |
| 3 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 3qh3_A-B_tcr.pdb | CDR-B1 | 1 | beta_chain | 5.0 | 0.319232 |
| 4 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 3qh3_A-B_tcr.pdb | CDR-B2 | 2 | beta_chain | 6.0 | 0.167444 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 569 | 7r7z_A-B_tcr.pdb | 7r80_A-B-E-C-D_tcr_pmhc.pdb | CDR-A2 | 2 | alpha_chain | 7.0 | 0.173771 |
| 570 | 7r7z_A-B_tcr.pdb | 7r80_A-B-E-C-D_tcr_pmhc.pdb | CDR-A3 | 3 | alpha_chain | 11.0 | 0.508615 |
| 571 | 7r7z_A-B_tcr.pdb | 7r80_A-B-E-C-D_tcr_pmhc.pdb | CDR-B1 | 1 | beta_chain | 5.0 | 0.108211 |
| 572 | 7r7z_A-B_tcr.pdb | 7r80_A-B-E-C-D_tcr_pmhc.pdb | CDR-B2 | 2 | beta_chain | 6.0 | 0.219891 |
| 573 | 7r7z_A-B_tcr.pdb | 7r80_A-B-E-C-D_tcr_pmhc.pdb | CDR-B3 | 3 | beta_chain | 13.0 | 0.789117 |
574 rows × 7 columns
[27]:
r, p_val = scipy.stats.pearsonr(results_tcr_apex['cdr_length'], results_tcr_apex['apex_ca_distance'])
sns.lmplot(results_tcr_apex.sort_values('cdr_type'), x='cdr_length', y='apex_ca_distance', scatter=False)
sns.scatterplot(results_tcr_apex.sort_values('cdr_type'), x='cdr_length', y='apex_ca_distance', hue='cdr_type')
plt.text(6.75, 4, f'$r^2$ = {r**2: .2f}, p-value = {p_val: .2e}')
plt.show()
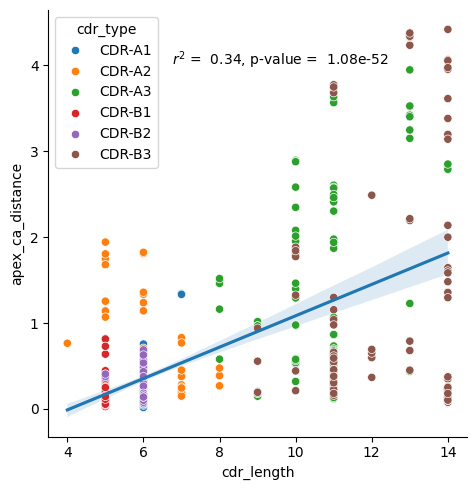
Peptide Analysis
Load data
[28]:
results_peptide = pd.read_csv(os.path.join(DATA_DIR, 'pmhc_tcr_contact_apo_holo.csv'))
results_peptide = results_peptide.query("chain_type == 'antigen_chain'")
results_peptide = results_peptide.drop(columns=['chain_type', 'tcr_contact'])
results_peptide
[28]:
| complex_id | structure_x_name | structure_y_name | rmsd | |
|---|---|---|---|---|
| 2 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | 0.448858 |
| 5 | 5wlg_D-E-C-A-B_tcr_pmhc | 5wlg_D-E-C-A-B_tcr_pmhc.pdb | 5wli_C-A-B_pmhc.pdb | 0.498148 |
| 8 | 5wlg_D-E-C-A-B_tcr_pmhc | 5wlg_D-E-C-A-B_tcr_pmhc.pdb | 5wli_F-D-E_pmhc.pdb | 0.519507 |
| 11 | 5wlg_D-E-C-A-B_tcr_pmhc | 5wlg_D-E-C-A-B_tcr_pmhc.pdb | 5wli_I-G-H_pmhc.pdb | 0.459025 |
| 14 | 5wlg_D-E-C-A-B_tcr_pmhc | 5wlg_D-E-C-A-B_tcr_pmhc.pdb | 5wli_L-J-K_pmhc.pdb | 0.494705 |
| ... | ... | ... | ... | ... |
| 3301 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n6d_O-M-N_pmhc.pdb | 7rtd_C-A-B_pmhc.pdb | 0.486296 |
| 3304 | 7rtr_D-E-C-A-B_tcr_pmhc | 7n6d_O-M-N_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 0.448905 |
| 3307 | 7rtr_D-E-C-A-B_tcr_pmhc | 7p3d_C-A-B_pmhc.pdb | 7rtd_C-A-B_pmhc.pdb | 0.773692 |
| 3310 | 7rtr_D-E-C-A-B_tcr_pmhc | 7p3d_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 0.765295 |
| 3313 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 0.455078 |
1096 rows × 4 columns
Merge with metadata
[29]:
apo_holo_summary_df['peptide_length'] = apo_holo_summary_df['peptide_sequence'].str.len()
[30]:
results_peptide = results_peptide.merge(
apo_holo_summary_df[['file_name', 'pdb_id', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary_df[['file_name', 'pdb_id', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
).merge(
apo_holo_summary_df[['id', 'peptide_sequence', 'peptide_length', 'mhc_slug']],
how='left',
left_on='complex_id',
right_on='id',
)
Normalise data
[31]:
results_peptide['comparison'] = results_peptide['state_x'] + '-' + results_peptide['state_y']
results_peptide['comparison'] = results_peptide['comparison'].map(
lambda entry: 'apo-holo' if entry == 'holo-apo' else entry,
)
results_peptide = results_peptide.query("comparison == 'apo-holo'").reset_index(drop=True)
[32]:
results_peptide['structure_comparison'] = results_peptide.apply(
lambda row: '-'.join(sorted([row.structure_x_name, row.structure_y_name])),
axis='columns',
)
results_peptide = results_peptide.drop_duplicates('structure_comparison')
[33]:
results_peptide = results_peptide.groupby(['peptide_sequence',
'comparison',
'peptide_length',
'mhc_slug'])['rmsd'].mean().reset_index()
[34]:
results_peptide
[34]:
| peptide_sequence | comparison | peptide_length | mhc_slug | rmsd | |
|---|---|---|---|---|---|
| 0 | AAGIGILTV | apo-holo | 9.0 | hla_a_02_01 | 1.025622 |
| 1 | ALGIGILTV | apo-holo | 9.0 | hla_a_02_01 | 1.274516 |
| 2 | ALWGFFPVL | apo-holo | 9.0 | hla_a_02_01 | 0.351748 |
| 3 | ALWGPDPAAA | apo-holo | 10.0 | hla_a_02_01 | 0.703094 |
| 4 | APRGPHGGAASGL | apo-holo | 13.0 | hla_b_07_02 | 3.523593 |
| ... | ... | ... | ... | ... | ... |
| 75 | VVVGAGGVGK | apo-holo | 10.0 | hla_a_11_01 | 1.483028 |
| 76 | YGFRNVVHI | apo-holo | 9.0 | h2_db | 0.275006 |
| 77 | YLGGPDFPTI | apo-holo | 10.0 | hla_a_02_01 | 0.868938 |
| 78 | YLQPRTFLL | apo-holo | 9.0 | hla_a_02_01 | 0.584577 |
| 79 | YQFGPDFPIA | apo-holo | 10.0 | hla_a_02_01 | 0.408370 |
80 rows × 5 columns
Visualise results
[35]:
r, p_val = scipy.stats.pearsonr(results_peptide['peptide_length'], results_peptide['rmsd'])
sns.lmplot(results_peptide.sort_values('mhc_slug'), x='peptide_length', y='rmsd', scatter=False)
ax = sns.scatterplot(results_peptide.sort_values('mhc_slug'), x='peptide_length', y='rmsd', hue='mhc_slug')
sns.move_legend(ax, "upper left", bbox_to_anchor=(1, 1))
plt.text(8.5, 4, f'$r^2$ = {r**2: .2f}, p-value = {p_val: .2e}')
plt.show()
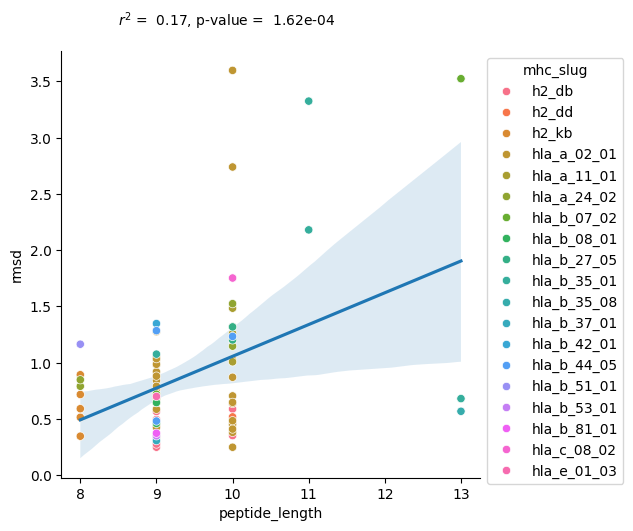
Conclusion
Both TCR CDR loops and peptides show a correlation between length and amount of conformational change. For the TCRs, there is a correlation in both paradigms, framework alignment and loop alignment, but the correlation is much stronger looking at the loop alignments. The increased correlation for loop alignments make sense since the conformational changes from the framework regions can be driven by other parts of the protein, but when the loops are aligned together, the only differences can be driven by changes in the loops themselves, implying the loop length has more of an effect.