pMHC movement between apo and holo conformations
Introduction
In this notebook we aim to determine what parts of the pMHC, if any, move when contacted by TCRs. We split the MHC domain into MHC-non-TCR contact and MHC-TCR contact based on previous analysis of the TCR finger print on the MHC molecules.
[1]:
import itertools
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scipy
import seaborn as sns
[2]:
NOISE_LEVEL = 0.5 # Å
Load comparisons
[3]:
results = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I-comparisons/pmhc_tcr_contact_apo_holo.csv')
results
[3]:
| complex_id | structure_x_name | structure_y_name | chain_type | tcr_contact | rmsd | |
|---|---|---|---|---|---|---|
| 0 | 3qdg_D-E-C-A-B_tcr_pmhc | 1jf1_C-A-B_pmhc.pdb | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | mhc_chain1 | False | 0.630013 |
| 1 | 3qdg_D-E-C-A-B_tcr_pmhc | 1jf1_C-A-B_pmhc.pdb | 3qdg_D-E-C-A-B_tcr_pmhc.pdb | mhc_chain1 | True | 0.582077 |
| 2 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | antigen_chain | False | 0.448858 |
| 3 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | mhc_chain1 | False | 0.422160 |
| 4 | 5c0a_D-E-C-A-B_tcr_pmhc | 5c0a_D-E-C-A-B_tcr_pmhc.pdb | 5n1y_C-A-B_pmhc.pdb | mhc_chain1 | True | 0.424287 |
| ... | ... | ... | ... | ... | ... | ... |
| 3311 | 7rtr_D-E-C-A-B_tcr_pmhc | 7p3d_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | mhc_chain1 | False | 0.497769 |
| 3312 | 7rtr_D-E-C-A-B_tcr_pmhc | 7p3d_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | mhc_chain1 | True | 0.458372 |
| 3313 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | antigen_chain | False | 0.455078 |
| 3314 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | mhc_chain1 | False | 0.449558 |
| 3315 | 7rtr_D-E-C-A-B_tcr_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | mhc_chain1 | True | 0.493225 |
3316 rows × 6 columns
[4]:
results_holo_holo = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I-comparisons/pmhc_tcr_contact_holo.csv')
[5]:
results_holo_holo = results_holo_holo.query("chain_type == 'mhc_chain1' or chain_type == 'antigen_chain'")
[6]:
results_holo_holo['mhc_slug'] = None
results_holo_holo['peptide_sequence'] = None
[7]:
mhc_pattern = r'^hla|h2'
mhc_complex_ids = results_holo_holo['complex_id'].str.contains(mhc_pattern, regex=True)
mhc_slug_peptides = results_holo_holo[mhc_complex_ids]['complex_id'].str.rsplit('_', n=1)
mhc_slugs = mhc_slug_peptides.map(lambda composite: composite[0])
peptides = mhc_slug_peptides.map(lambda composite: composite[1])
results_holo_holo.loc[mhc_complex_ids, 'mhc_slug'] = mhc_slugs
results_holo_holo.loc[mhc_complex_ids, 'peptide_sequence'] = peptides
results_holo_holo = results_holo_holo[mhc_complex_ids]
Load summary data
[8]:
apo_holo_summary_df = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I/apo_holo_summary.csv')
apo_holo_summary_df
[8]:
| file_name | pdb_id | structure_type | state | alpha_chain | beta_chain | antigen_chain | mhc_chain1 | mhc_chain2 | cdr_sequences_collated | peptide_sequence | mhc_slug | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 1ao7 | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVTTDSWGKLQ-MNHEY-SVGAGI-ASRPGLA... | LLFGYPVYV | hla_a_02_01 |
| 1 | 1b0g_C-A-B_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | C | A | B | NaN | ALWGFFPVL | hla_a_02_01 |
| 2 | 1b0g_F-D-E_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | F | D | E | NaN | ALWGFFPVL | hla_a_02_01 |
| 3 | 1bd2_D-E-C-A-B_tcr_pmhc.pdb | 1bd2 | tcr_pmhc | holo | D | E | C | A | B | NSMFDY-ISSIKDK-AAMEGAQKLV-MNHEY-SVGAGI-ASSYPGG... | LLFGYPVYV | hla_a_02_01 |
| 4 | 1bii_P-A-B_pmhc.pdb | 1bii | pmhc | apo | NaN | NaN | P | A | B | NaN | RGPGRAFVTI | h2_dd |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 386 | 7rtd_C-A-B_pmhc.pdb | 7rtd | pmhc | apo | NaN | NaN | C | A | B | NaN | YLQPRTFLL | hla_a_02_01 |
| 387 | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 7rtr | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 |
| 388 | 8gvb_A-B-P-H-L_tcr_pmhc.pdb | 8gvb | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RYPLTFGW | hla_a_24_02 |
| 389 | 8gvg_A-B-P-H-L_tcr_pmhc.pdb | 8gvg | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RFPLTFGW | hla_a_24_02 |
| 390 | 8gvi_A-B-P-H-L_tcr_pmhc.pdb | 8gvi | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVVFTGGGNKLT-SEHNR-FQNEAQ-ASSL... | RYPLTFGW | hla_a_24_02 |
391 rows × 12 columns
[9]:
apo_holo_summary_df['ids'] = apo_holo_summary_df['file_name'].str.replace('.pdb$', '')
/var/scratch/bmcmaste/2178229/ipykernel_2205981/477882467.py:1: FutureWarning: The default value of regex will change from True to False in a future version.
apo_holo_summary_df['ids'] = apo_holo_summary_df['file_name'].str.replace('.pdb$', '')
Annotate results with summary data
[10]:
results = results.merge(apo_holo_summary_df[['ids', 'cdr_sequences_collated', 'peptide_sequence', 'mhc_slug']],
left_on='complex_id',
right_on='ids',
how='left')
[11]:
results = pd.concat([results, results_holo_holo])
[12]:
results = results.merge(
apo_holo_summary_df[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary_df[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
)
[13]:
def name_domain(chain_type: str, tcr_contact: bool) -> str | None:
match chain_type:
case 'antigen_chain':
return 'peptide'
case 'mhc_chain1':
if tcr_contact:
return 'mhc_tcr_contact'
return 'mhc'
return None
results['domain'] = results.apply(lambda row: name_domain(row.chain_type, row.tcr_contact), axis='columns')
[14]:
results['comparison'] = results['state_x'] + '-' + results['state_y']
results['comparison'] = results['comparison'].map(lambda entry: 'apo-holo' if entry == 'holo-apo' else entry)
[15]:
results['structure_comparison'] = results.apply(
lambda row: '-'.join(sorted([row.structure_x_name, row.structure_y_name])),
axis='columns',
)
results = results.drop_duplicates(['structure_comparison', 'domain'])
[16]:
results = results.groupby(['peptide_sequence',
'mhc_slug',
'comparison',
'domain'], dropna=False)['rmsd'].mean().reset_index()
[17]:
apo_holo_results = results.query("comparison == 'apo-holo'")
[18]:
apo_holo_results
[18]:
| peptide_sequence | mhc_slug | comparison | domain | rmsd | |
|---|---|---|---|---|---|
| 3 | AAGIGILTV | hla_a_02_01 | apo-holo | mhc | 0.660376 |
| 4 | AAGIGILTV | hla_a_02_01 | apo-holo | mhc_tcr_contact | 0.562211 |
| 5 | AAGIGILTV | hla_a_02_01 | apo-holo | peptide | 1.025622 |
| 12 | ALGIGILTV | hla_a_02_01 | apo-holo | mhc | 0.620830 |
| 13 | ALGIGILTV | hla_a_02_01 | apo-holo | mhc_tcr_contact | 0.554737 |
| ... | ... | ... | ... | ... | ... |
| 462 | YLQPRTFLL | hla_a_02_01 | apo-holo | mhc_tcr_contact | 0.578509 |
| 463 | YLQPRTFLL | hla_a_02_01 | apo-holo | peptide | 0.584577 |
| 467 | YQFGPDFPIA | hla_a_02_01 | apo-holo | mhc | 0.478941 |
| 468 | YQFGPDFPIA | hla_a_02_01 | apo-holo | mhc_tcr_contact | 0.357204 |
| 469 | YQFGPDFPIA | hla_a_02_01 | apo-holo | peptide | 0.408370 |
242 rows × 5 columns
Visualise results
[19]:
sns.boxplot(apo_holo_results, x='domain', y='rmsd')
x = np.linspace(-0.75, 2.75)
y = np.repeat(NOISE_LEVEL, len(x))
plt.plot(x, y, '--r')
plt.xlim(-0.75, 2.75)
[19]:
(-0.75, 2.75)
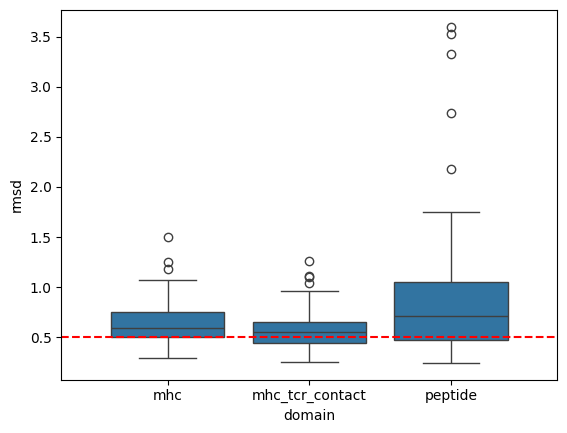
Compute Statistics
[20]:
sns.displot(apo_holo_results, x='rmsd', col='domain')
[20]:
<seaborn.axisgrid.FacetGrid at 0x7f2641194970>
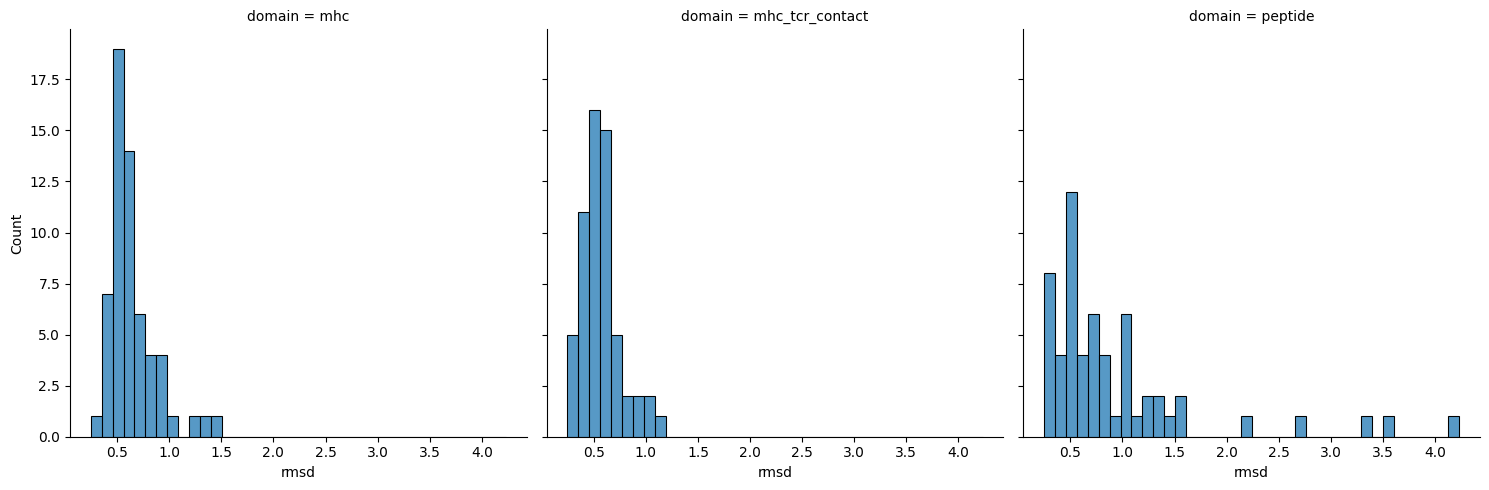
[21]:
conditions = {domain: apo_holo_results[apo_holo_results['domain'] == domain]['rmsd'].values for domain in apo_holo_results['domain'].unique()}
[22]:
scipy.stats.f_oneway(*conditions.values())
[22]:
F_onewayResult(statistic=13.305204363913258, pvalue=3.320938947108907e-06)
[23]:
combos = list(itertools.combinations(conditions.items(), 2))
significane_level = 0.05 / len(combos)
ad_hoc_results = {'condition_x': [], 'condition_y': [], 'stat': [], 'p_val': []}
for (condition_x_name, condition_x), (condition_y_name, condition_y) in combos:
ad_hoc_results['condition_x'].append(condition_x_name)
ad_hoc_results['condition_y'].append(condition_y_name)
stat, p_val = scipy.stats.ttest_ind(condition_x, condition_y)
ad_hoc_results['stat'].append(stat)
ad_hoc_results['p_val'].append(p_val)
ad_hoc_results = pd.DataFrame(ad_hoc_results)
ad_hoc_results['significant'] = ad_hoc_results['p_val'] < significane_level
ad_hoc_results
[23]:
| condition_x | condition_y | stat | p_val | significant | |
|---|---|---|---|---|---|
| 0 | mhc | mhc_tcr_contact | 2.391609 | 0.017935 | False |
| 1 | mhc | peptide | -3.203906 | 0.001638 | True |
| 2 | mhc_tcr_contact | peptide | -4.201672 | 0.000044 | True |
Based on these results it seems that the peptide undergoes conformational change but not the MHC molecule- either TCR-contacting or not.
Comparing the differences in binding between movement types
[24]:
sns.catplot(results,
x='comparison', y='rmsd',
col='domain',
kind='box')
[24]:
<seaborn.axisgrid.FacetGrid at 0x7f263ee23370>
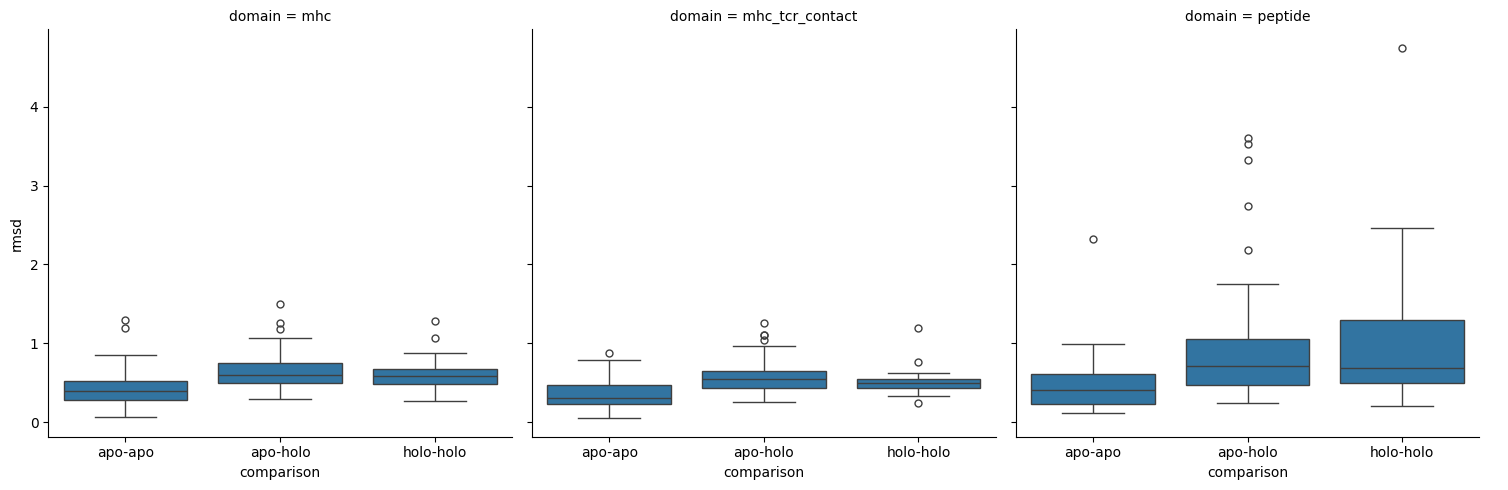
[25]:
sns.displot(results,
hue='comparison', x='rmsd',
col='domain',
kind='kde')
[25]:
<seaborn.axisgrid.FacetGrid at 0x7f263ee23fa0>
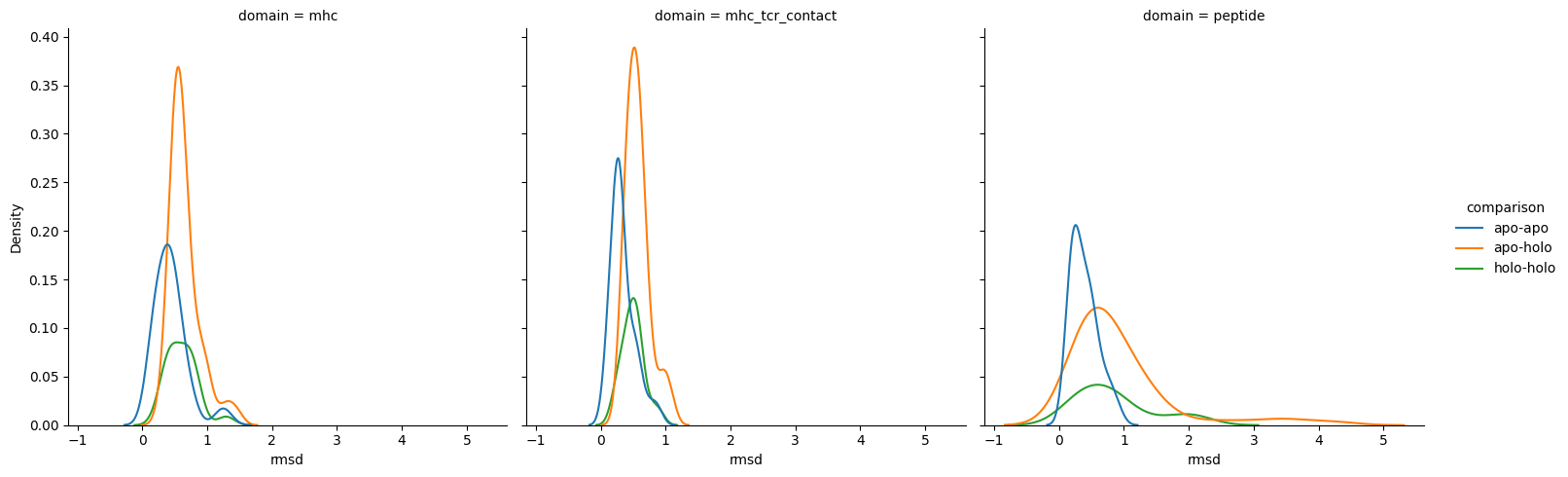
[26]:
treatment_options = ['comparison', 'domain']
treatments = [(group, df['rmsd'].to_numpy()) for group, df in results.groupby(treatment_options)]
treatments
print(scipy.stats.kruskal(*[values for _, values in treatments]))
KruskalResult(statistic=114.03107622089692, pvalue=5.640784471619738e-21)
[27]:
combos = []
for pairing in list(itertools.combinations(treatments, 2)):
domain_x = pairing[0][0][1]
domain_y = pairing[1][0][1]
if domain_x == domain_y:
combos.append(pairing)
significance_level = 0.05 / len(combos)
print(significance_level)
statistics = []
p_vals = []
for ((comparison_x, domain_x), sample_x), ((comparison_y, domain_y), sample_y) in combos:
stat, p_val = scipy.stats.ranksums(sample_x, sample_y)
statistics.append(stat)
p_vals.append(p_val)
momvement_type_statistics = pd.DataFrame({
'comparison_x': [name for ((name, _,), _), _ in combos],
'domain_x': [name for ((_, name), _), _ in combos],
'comparison_y': [name for _, ((name, _), _) in combos],
'domain_y': [name for _, ((_, name), _) in combos],
'statistic': statistics,
'p_val': p_vals,
'significant': [p_val < significance_level for p_val in p_vals],
})
momvement_type_statistics['p_val'] = momvement_type_statistics['p_val'].map(lambda num: f'{num:.2e}')
momvement_type_statistics
0.005555555555555556
[27]:
| comparison_x | domain_x | comparison_y | domain_y | statistic | p_val | significant | |
|---|---|---|---|---|---|---|---|
| 0 | apo-apo | mhc | apo-holo | mhc | -5.543219 | 2.97e-08 | True |
| 1 | apo-apo | mhc | holo-holo | mhc | -3.284693 | 1.02e-03 | True |
| 2 | apo-apo | mhc_tcr_contact | apo-holo | mhc_tcr_contact | -6.324955 | 2.53e-10 | True |
| 3 | apo-apo | mhc_tcr_contact | holo-holo | mhc_tcr_contact | -4.029224 | 5.60e-05 | True |
| 4 | apo-apo | peptide | apo-holo | peptide | -5.153956 | 2.55e-07 | True |
| 5 | apo-apo | peptide | holo-holo | peptide | -3.624112 | 2.90e-04 | True |
| 6 | apo-holo | mhc | holo-holo | mhc | 0.602876 | 5.47e-01 | False |
| 7 | apo-holo | mhc_tcr_contact | holo-holo | mhc_tcr_contact | 1.496293 | 1.35e-01 | False |
| 8 | apo-holo | peptide | holo-holo | peptide | -0.102800 | 9.18e-01 | False |
Conclusion
This analysis reveals that the peptide undergoes the most change from apo to holo states and the MHC molecule has little change between these states. It does not matter whether it is the TCR contacting portion of the MHC or not. The statistical analysis comparing the differences between comparion types gives inconclusive results.