Compare D-scores
Introduction
In this notebook, we analyze the apo-holo conformational changes of TCRs and pMHCs using the D-score derived from \(\phi\)- and \(\psi\)-angles. We aim to confirm are previous results that all CDR loops undergo conformational changes but only CDR3 loops are flexible, and further probe the underlying sources of the conformational changes.
D-score
Described in Mardia & Jupp, 2000 (Directional Statistics), the D-score measures the changes in backbone dihedral angles.
\(D(\theta_1, \theta_2) = 2(1 - \cos{(\theta_1 - \theta_2)})\)
\(\text{D-score}(A, B) = \sum_{i}^{n}(D(\phi_{i}^{A}, \phi_{i}^{B}) + D(\psi_{i}^{A}, \psi_{i}^{B}))\)
[1]:
import os
import itertools
import pandas as pd
import scipy
import seaborn as sns
from tcr_pmhc_interface_analysis.imgt_numbering import IMGT_CDR
[2]:
DATA_DIR = '../data/processed/apo-holo-tcr-pmhc-class-I-comparisons'
Load metadata
[3]:
apo_holo_summary = pd.read_csv('../data/processed/apo-holo-tcr-pmhc-class-I/apo_holo_summary.csv')
apo_holo_summary['id'] = apo_holo_summary['file_name'].str.replace('.pdb', '', regex=False)
apo_holo_summary = apo_holo_summary.set_index('id')
apo_holo_summary
[3]:
| file_name | pdb_id | structure_type | state | alpha_chain | beta_chain | antigen_chain | mhc_chain1 | mhc_chain2 | cdr_sequences_collated | peptide_sequence | mhc_slug | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||
| 1ao7_D-E-C-A-B_tcr_pmhc | 1ao7_D-E-C-A-B_tcr_pmhc.pdb | 1ao7 | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVTTDSWGKLQ-MNHEY-SVGAGI-ASRPGLA... | LLFGYPVYV | hla_a_02_01 |
| 1b0g_C-A-B_pmhc | 1b0g_C-A-B_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | C | A | B | NaN | ALWGFFPVL | hla_a_02_01 |
| 1b0g_F-D-E_pmhc | 1b0g_F-D-E_pmhc.pdb | 1b0g | pmhc | apo | NaN | NaN | F | D | E | NaN | ALWGFFPVL | hla_a_02_01 |
| 1bd2_D-E-C-A-B_tcr_pmhc | 1bd2_D-E-C-A-B_tcr_pmhc.pdb | 1bd2 | tcr_pmhc | holo | D | E | C | A | B | NSMFDY-ISSIKDK-AAMEGAQKLV-MNHEY-SVGAGI-ASSYPGG... | LLFGYPVYV | hla_a_02_01 |
| 1bii_P-A-B_pmhc | 1bii_P-A-B_pmhc.pdb | 1bii | pmhc | apo | NaN | NaN | P | A | B | NaN | RGPGRAFVTI | h2_dd |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 7rtd_C-A-B_pmhc | 7rtd_C-A-B_pmhc.pdb | 7rtd | pmhc | apo | NaN | NaN | C | A | B | NaN | YLQPRTFLL | hla_a_02_01 |
| 7rtr_D-E-C-A-B_tcr_pmhc | 7rtr_D-E-C-A-B_tcr_pmhc.pdb | 7rtr | tcr_pmhc | holo | D | E | C | A | B | DRGSQS-IYSNGD-AVNRDDKII-SEHNR-FQNEAQ-ASSPDIEQY | YLQPRTFLL | hla_a_02_01 |
| 8gvb_A-B-P-H-L_tcr_pmhc | 8gvb_A-B-P-H-L_tcr_pmhc.pdb | 8gvb | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RYPLTFGW | hla_a_24_02 |
| 8gvg_A-B-P-H-L_tcr_pmhc | 8gvg_A-B-P-H-L_tcr_pmhc.pdb | 8gvg | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVGFTGGGNKLT-SEHNR-FQNEAQ-ASSD... | RFPLTFGW | hla_a_24_02 |
| 8gvi_A-B-P-H-L_tcr_pmhc | 8gvi_A-B-P-H-L_tcr_pmhc.pdb | 8gvi | tcr_pmhc | holo | A | B | P | H | L | YGATPY-YFSGDTLV-AVVFTGGGNKLT-SEHNR-FQNEAQ-ASSL... | RYPLTFGW | hla_a_24_02 |
391 rows × 12 columns
Analysis of TCR D-scores
Load TCR data
[4]:
tcr_d_score_df = pd.read_csv(os.path.join(DATA_DIR, 'tcr_per_res_apo_holo_d_score.csv'))
[5]:
tcr_d_score_df = tcr_d_score_df.merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
).merge(
apo_holo_summary[['cdr_sequences_collated', 'peptide_sequence', 'mhc_slug']],
how='left',
left_on='complex_id',
right_index=True,
)
[6]:
d_score_df_holo = pd.read_csv(os.path.join(DATA_DIR, 'tcr_per_res_holo_holo_d_score.csv'))
d_score_df_holo['cdr_sequences_collated'] = None
cdr_pattern = r'^[A-Z]+-[A-Z]+-[A-Z]+-[A-Z]+-[A-Z]+-[A-Z]+'
tcr_aligned_complex_ids = d_score_df_holo['complex_id'].str.contains(cdr_pattern)
holo_cdr_sequences_collated = d_score_df_holo[tcr_aligned_complex_ids]['complex_id']
d_score_df_holo.loc[tcr_aligned_complex_ids, 'cdr_sequences_collated'] = holo_cdr_sequences_collated
d_score_df_holo = d_score_df_holo.merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
)
d_score_df_holo_tcr = d_score_df_holo[tcr_aligned_complex_ids]
[7]:
tcr_d_score_df = pd.concat([tcr_d_score_df, d_score_df_holo_tcr])
[8]:
tcr_d_score_df['comparison'] = tcr_d_score_df['state_x'] + '-' + tcr_d_score_df['state_y']
tcr_d_score_df['comparison'] = tcr_d_score_df['comparison'].map(
lambda entry: 'apo-holo' if entry == 'holo-apo' else entry
)
[9]:
tcr_d_score_df['structure_comparison'] = tcr_d_score_df.apply(
lambda row: '-'.join(sorted([row.structure_x_name, row.structure_y_name])),
axis='columns',
)
tcr_d_score_df = tcr_d_score_df.drop_duplicates(['structure_comparison', 'chain_type', 'cdr',
'residue_name', 'residue_seq_id', 'residue_insert_code'])
tcr_d_score_df = tcr_d_score_df.reset_index(drop=True)
[10]:
tcr_d_score_df = tcr_d_score_df[~tcr_d_score_df['d_score'].isnull()].reset_index(drop=True)
[11]:
tcr_d_score_df = tcr_d_score_df.groupby(['cdr_sequences_collated',
'comparison',
'chain_type',
'cdr',
'residue_name',
'residue_seq_id',
'residue_insert_code'], dropna=False)['d_score'].apply('mean').reset_index()
[12]:
tcr_d_score_df['anchor'] = tcr_d_score_df['residue_seq_id'].map(lambda seq_id: seq_id not in IMGT_CDR)
[13]:
tcr_d_score_df
[13]:
| cdr_sequences_collated | comparison | chain_type | cdr | residue_name | residue_seq_id | residue_insert_code | d_score | anchor | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | ATGYPS-ATKADDK-ALSDPVNDMR-SGHAT-FQNNGV-ASSLRGR... | apo-holo | alpha_chain | 1 | ALA | 27 | NaN | 0.138574 | False |
| 1 | ATGYPS-ATKADDK-ALSDPVNDMR-SGHAT-FQNNGV-ASSLRGR... | apo-holo | alpha_chain | 1 | ASN | 22 | NaN | 0.076004 | True |
| 2 | ATGYPS-ATKADDK-ALSDPVNDMR-SGHAT-FQNNGV-ASSLRGR... | apo-holo | alpha_chain | 1 | CYS | 23 | NaN | 0.009379 | True |
| 3 | ATGYPS-ATKADDK-ALSDPVNDMR-SGHAT-FQNNGV-ASSLRGR... | apo-holo | alpha_chain | 1 | GLY | 29 | NaN | 3.322146 | False |
| 4 | ATGYPS-ATKADDK-ALSDPVNDMR-SGHAT-FQNNGV-ASSLRGR... | apo-holo | alpha_chain | 1 | LEU | 39 | NaN | 0.073606 | True |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 6526 | YSGSPE-HISR-ALSGFNNAGNMLT-SGHAT-FQNNGV-ASSLGGA... | apo-holo | beta_chain | 3 | THR | 115 | NaN | 0.000685 | False |
| 6527 | YSGSPE-HISR-ALSGFNNAGNMLT-SGHAT-FQNNGV-ASSLGGA... | apo-holo | beta_chain | 3 | THR | 122 | NaN | 0.192570 | True |
| 6528 | YSGSPE-HISR-ALSGFNNAGNMLT-SGHAT-FQNNGV-ASSLGGA... | apo-holo | beta_chain | 3 | TYR | 102 | NaN | 0.003136 | True |
| 6529 | YSGSPE-HISR-ALSGFNNAGNMLT-SGHAT-FQNNGV-ASSLGGA... | apo-holo | beta_chain | 3 | TYR | 117 | NaN | 0.032772 | False |
| 6530 | YSGSPE-HISR-ALSGFNNAGNMLT-SGHAT-FQNNGV-ASSLGGA... | apo-holo | beta_chain | 3 | VAL | 101 | NaN | 0.009630 | True |
6531 rows × 9 columns
Analysis
Comparing apo-apo, apo-holo, and holo-holo D-scores for the CDR Loops
[14]:
cdr_d_scores = (tcr_d_score_df.query('not anchor')
.groupby(['cdr_sequences_collated', 'comparison', 'chain_type', 'cdr'],
dropna=False)['d_score']
.apply('sum')
.reset_index())
[15]:
cdr_d_scores['similar'] = cdr_d_scores['d_score'] <= 1.5
[16]:
sns.catplot(cdr_d_scores.sort_values('comparison'),
row='chain_type', col='cdr',
x='comparison',
y='d_score',
kind='box')
[16]:
<seaborn.axisgrid.FacetGrid at 0x7fe420341030>
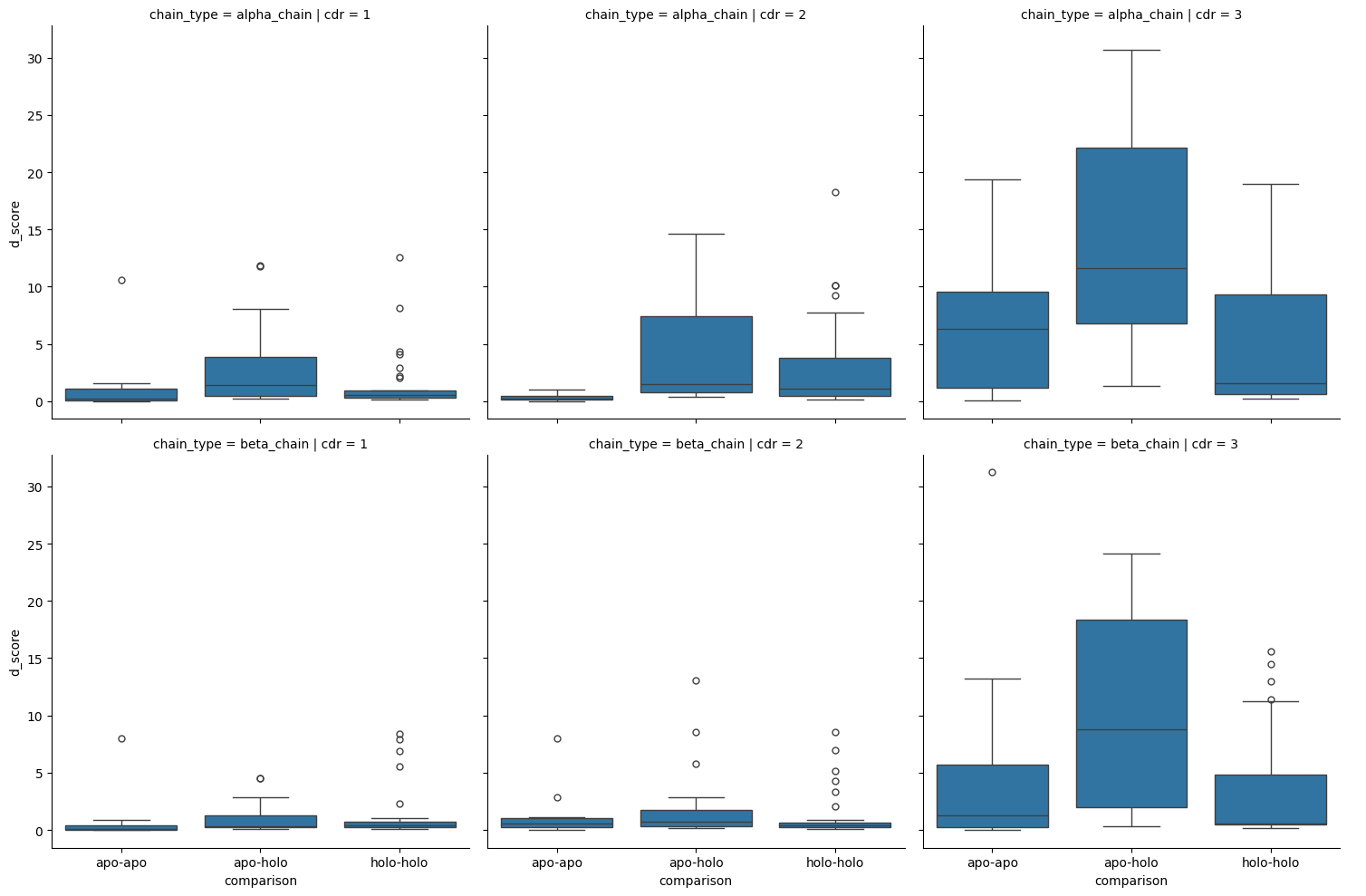
[17]:
sns.displot(cdr_d_scores,
row='chain_type', col='cdr',
hue='comparison',
x='d_score',
kind='kde')
[17]:
<seaborn.axisgrid.FacetGrid at 0x7fe3f2d07fd0>
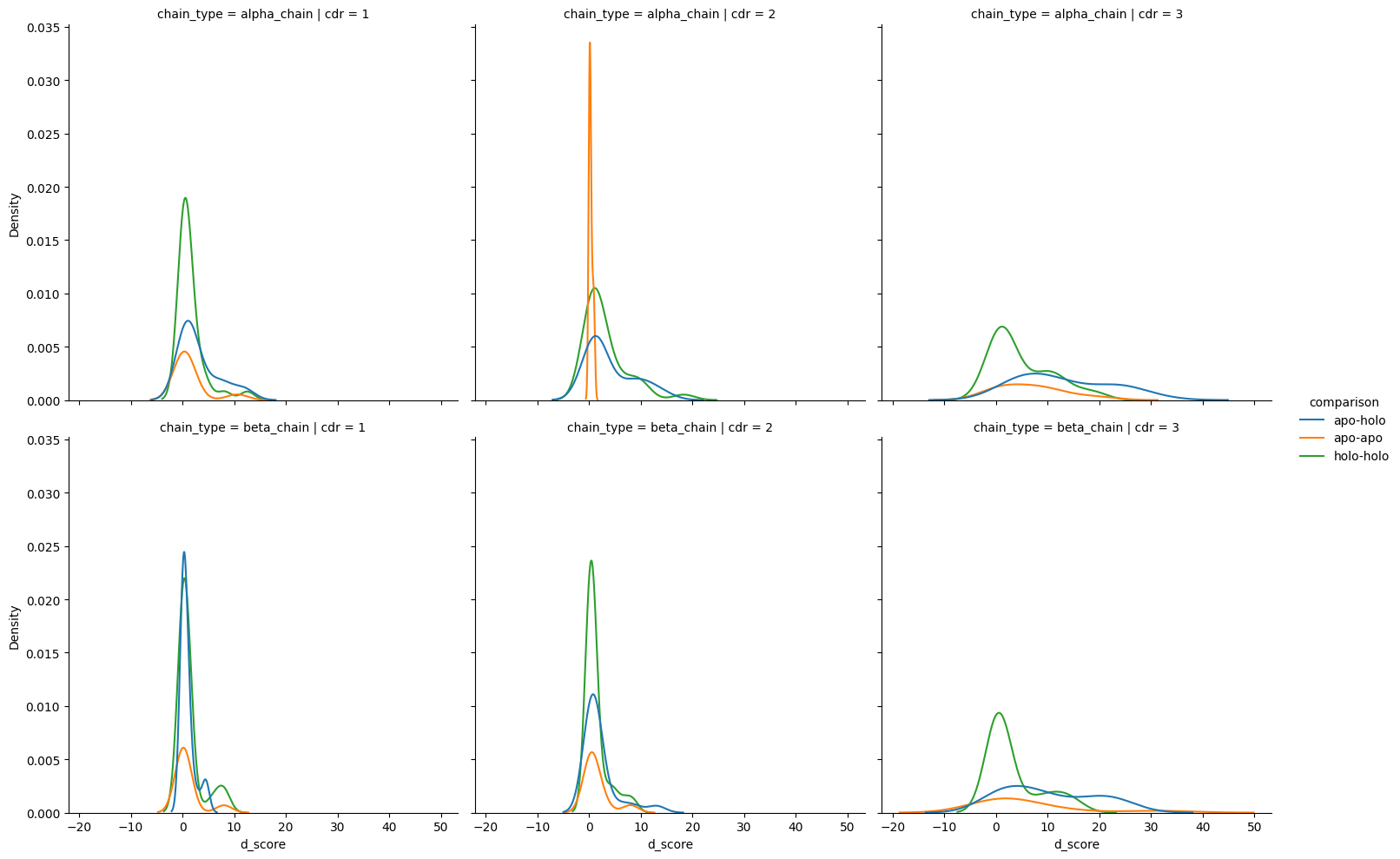
[18]:
treatment_options = ['comparison', 'chain_type', 'cdr']
treatments = [(group, df['d_score'].to_numpy()) for group, df in cdr_d_scores.groupby(treatment_options)]
treatments
print(scipy.stats.kruskal(*[values for _, values in treatments]))
KruskalResult(statistic=123.32856486268929, pvalue=3.571547724890704e-18)
[19]:
combos = []
for pairing in list(itertools.combinations(treatments, 2)):
chain_type_x = pairing[0][0][1]
cdr_x = pairing[0][0][2]
chain_type_y = pairing[1][0][1]
cdr_y = pairing[1][0][2]
if (chain_type_x, cdr_x) == (chain_type_y, cdr_y):
combos.append(pairing)
significance_level = 0.05 / len(combos)
print(significance_level)
statistics = []
p_vals = []
for ((comparison_x, chain_type_x, cdr_x), sample_x), ((comparison_y, chain_type_y, cdr_y), sample_y) in combos:
stat, p_val = scipy.stats.ranksums(sample_x, sample_y)
statistics.append(stat)
p_vals.append(p_val)
d_score_statistics_tcr = pd.DataFrame({
'comparison_x': [name for ((name, _, _), _), _ in combos],
'chain_type_x': [name for ((_, name, _), _), _ in combos],
'cdr_x': [name for ((_, _, name), _), _ in combos],
'comparison_y': [name for _, ((name, _, _), _) in combos],
'chain_type_y': [name for _, ((_, name, _), _) in combos],
'cdr_y': [name for _, ((_, _, name), _) in combos],
'statistic': statistics,
'p_val': p_vals,
'significant': [p_val < significance_level for p_val in p_vals],
})
d_score_statistics_tcr['p_val'] = d_score_statistics_tcr['p_val'].map(lambda num: f'{num:.2e}')
d_score_statistics_tcr
0.002777777777777778
[19]:
| comparison_x | chain_type_x | cdr_x | comparison_y | chain_type_y | cdr_y | statistic | p_val | significant | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | apo-apo | alpha_chain | 1 | apo-holo | alpha_chain | 1 | -2.450947 | 1.42e-02 | False |
| 1 | apo-apo | alpha_chain | 1 | holo-holo | alpha_chain | 1 | -1.811616 | 7.00e-02 | False |
| 2 | apo-apo | alpha_chain | 2 | apo-holo | alpha_chain | 2 | -3.887710 | 1.01e-04 | True |
| 3 | apo-apo | alpha_chain | 2 | holo-holo | alpha_chain | 2 | -3.404588 | 6.63e-04 | True |
| 4 | apo-apo | alpha_chain | 3 | apo-holo | alpha_chain | 3 | -1.910822 | 5.60e-02 | False |
| 5 | apo-apo | alpha_chain | 3 | holo-holo | alpha_chain | 3 | 0.437287 | 6.62e-01 | False |
| 6 | apo-apo | beta_chain | 1 | apo-holo | beta_chain | 1 | -1.870166 | 6.15e-02 | False |
| 7 | apo-apo | beta_chain | 1 | holo-holo | beta_chain | 1 | -2.061494 | 3.93e-02 | False |
| 8 | apo-apo | beta_chain | 2 | apo-holo | beta_chain | 2 | -0.935083 | 3.50e-01 | False |
| 9 | apo-apo | beta_chain | 2 | holo-holo | beta_chain | 2 | -0.093704 | 9.25e-01 | False |
| 10 | apo-apo | beta_chain | 3 | apo-holo | beta_chain | 3 | -1.992133 | 4.64e-02 | False |
| 11 | apo-apo | beta_chain | 3 | holo-holo | beta_chain | 3 | -0.124939 | 9.01e-01 | False |
| 12 | apo-holo | alpha_chain | 1 | holo-holo | alpha_chain | 1 | 2.009592 | 4.45e-02 | False |
| 13 | apo-holo | alpha_chain | 2 | holo-holo | alpha_chain | 2 | 1.186616 | 2.35e-01 | False |
| 14 | apo-holo | alpha_chain | 3 | holo-holo | alpha_chain | 3 | 3.685817 | 2.28e-04 | True |
| 15 | apo-holo | beta_chain | 1 | holo-holo | beta_chain | 1 | -0.407477 | 6.84e-01 | False |
| 16 | apo-holo | beta_chain | 2 | holo-holo | beta_chain | 2 | 1.296519 | 1.95e-01 | False |
| 17 | apo-holo | beta_chain | 3 | holo-holo | beta_chain | 3 | 3.556165 | 3.76e-04 | True |
Per-residue D-score of loop residues
[20]:
tcr_d_score_df['resi'] = tcr_d_score_df['residue_seq_id'].apply(str) + tcr_d_score_df['residue_insert_code'].fillna('')
[21]:
sns.catplot(tcr_d_score_df.query("comparison == 'apo-holo' and not anchor").sort_values(['resi', 'chain_type', 'cdr']),
row='chain_type', col='cdr',
x='resi', y='d_score',
sharex=False,
kind='bar')
[21]:
<seaborn.axisgrid.FacetGrid at 0x7fe3f331ce80>
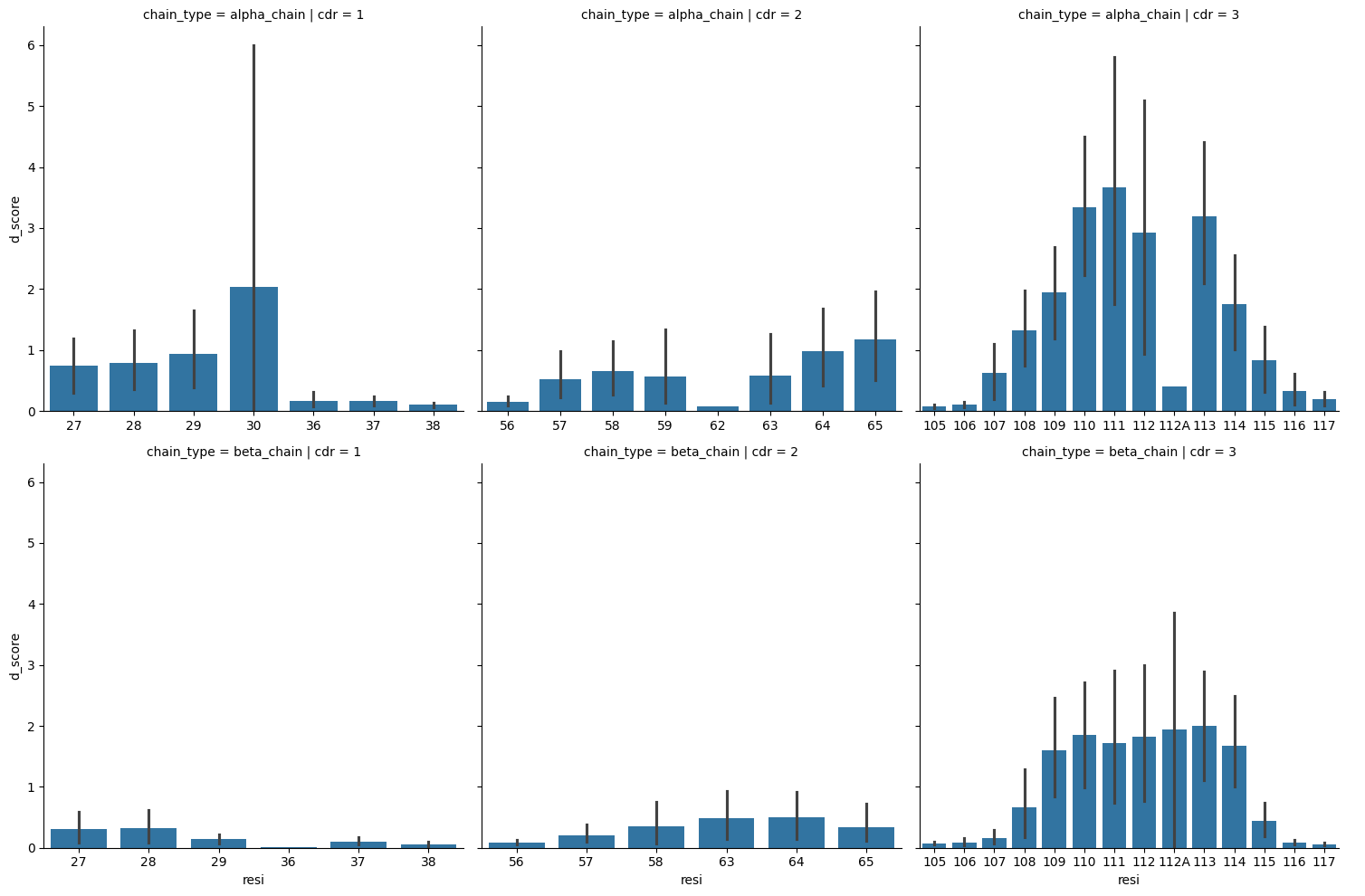
Comapring the D-scores of Loop residues versus anchor residues
[22]:
tcr_d_score_df['classification'] = tcr_d_score_df['anchor'].map(lambda anchor: 'anchor' if anchor else 'loop')
[23]:
apo_holo_loop_vs_anchor = (tcr_d_score_df.query("comparison == 'apo-holo'")
.groupby(['cdr_sequences_collated',
'chain_type', 'cdr',
'classification'],
dropna=False)['d_score']
.apply('sum')
.reset_index())
[24]:
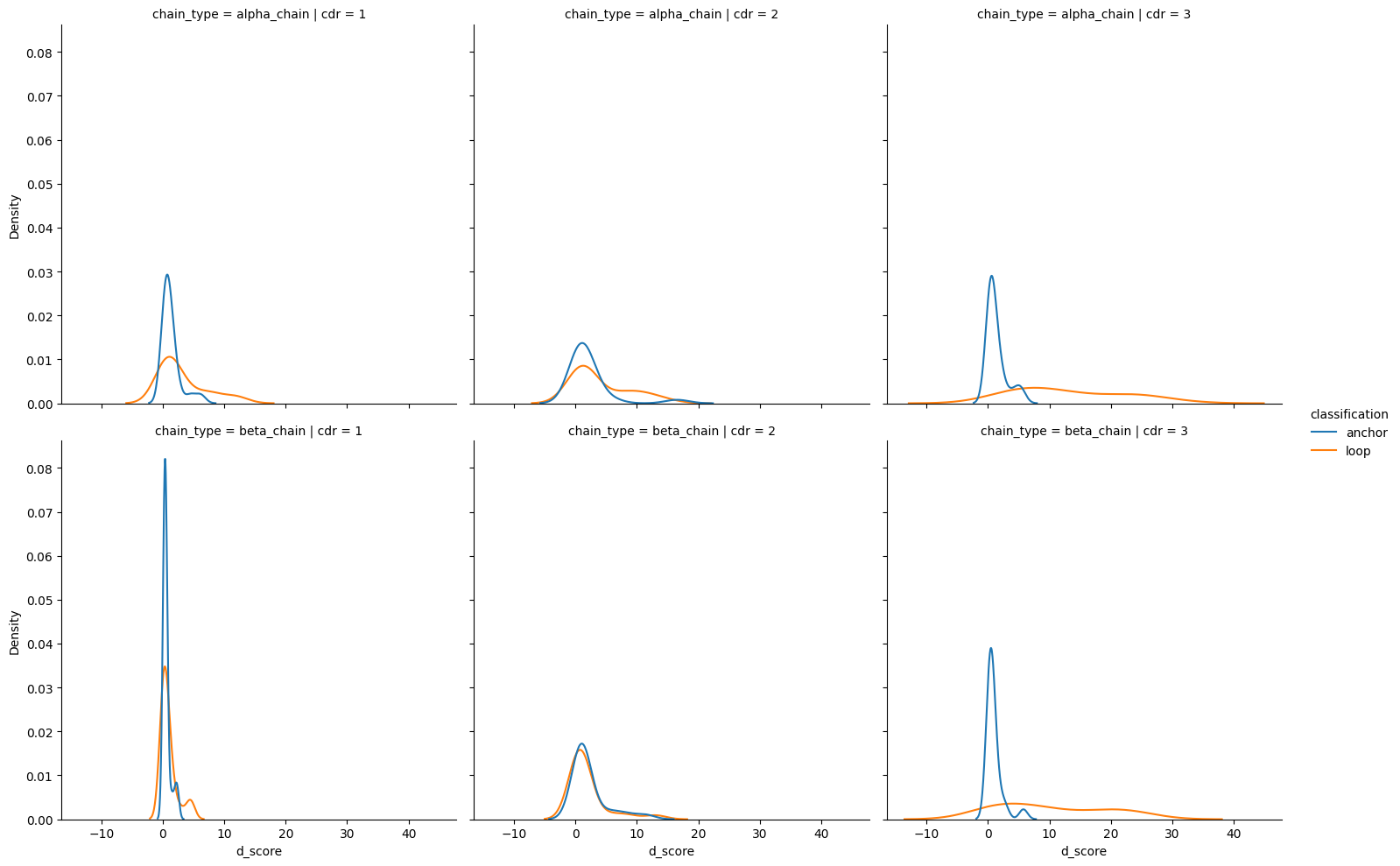
sns.catplot(apo_holo_loop_vs_anchor,
row='chain_type', col='cdr',
x='classification',
y='d_score',
kind='box')
[24]:
<seaborn.axisgrid.FacetGrid at 0x7fe3f2e12e30>
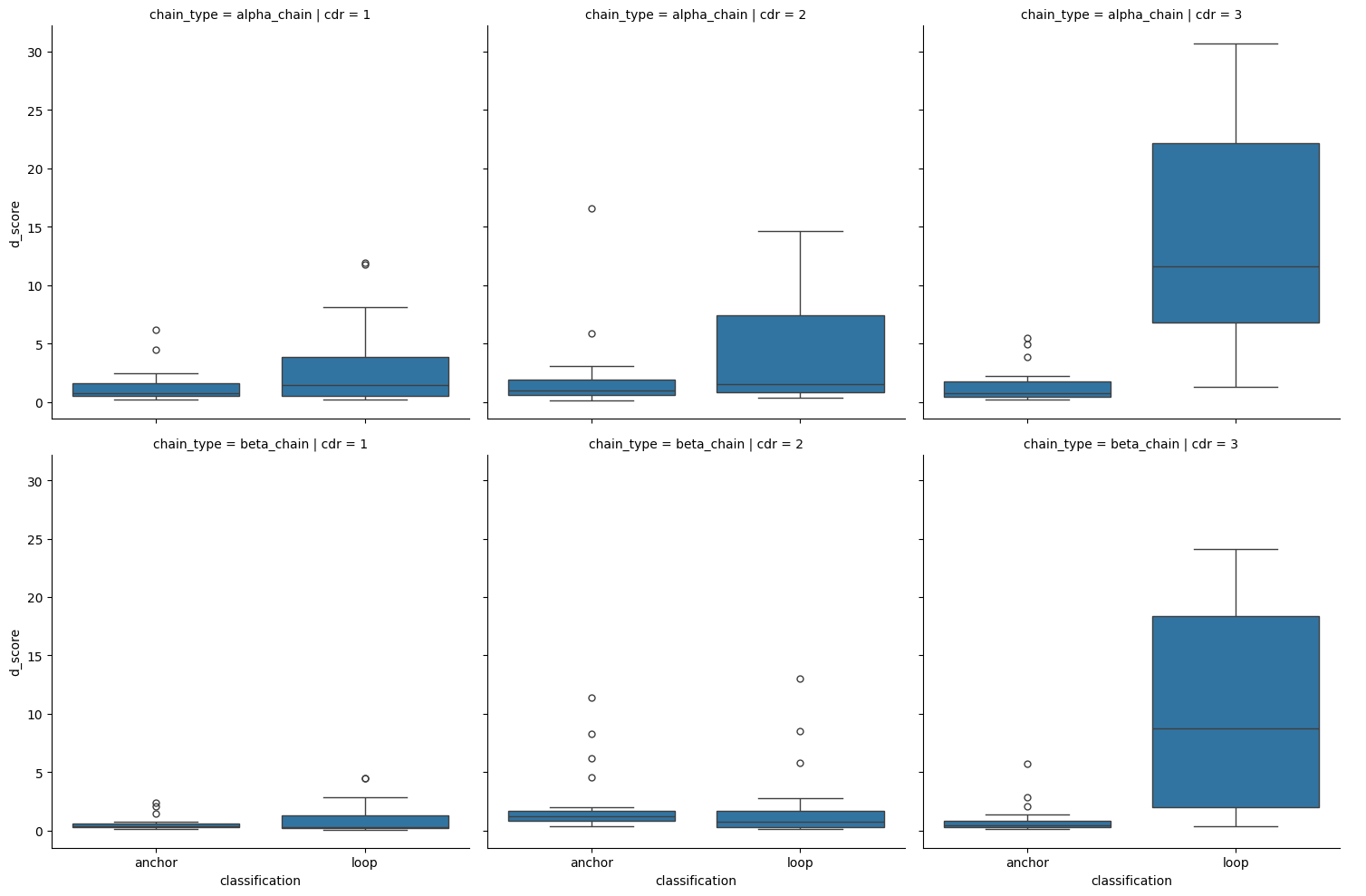
[25]:
sns.displot(apo_holo_loop_vs_anchor,
row='chain_type', col='cdr',
hue='classification',
x='d_score',
kind='kde')
[25]:
<seaborn.axisgrid.FacetGrid at 0x7fe3f0b9f910>
[26]:
treatment_options = ['classification', 'chain_type', 'cdr']
treatments = [(group, df['d_score'].to_numpy()) for group, df in apo_holo_loop_vs_anchor.groupby(treatment_options)]
print(scipy.stats.kruskal(*[values for _, values in treatments]))
KruskalResult(statistic=100.36714473266193, pvalue=1.5105310954050626e-16)
[27]:
combos = []
for pairing in list(itertools.combinations(treatments, 2)):
chain_type_x = pairing[0][0][1]
cdr_x = pairing[0][0][2]
chain_type_y = pairing[1][0][1]
cdr_y = pairing[1][0][2]
if (chain_type_x, cdr_x) == (chain_type_y, cdr_y):
combos.append(pairing)
significance_level = 0.05 / len(combos)
print(significance_level)
statistics = []
p_vals = []
for ((classification_x, chain_type_x, cdr_x), sample_x), ((classification_y, chain_type_y, cdr_y), sample_y) in combos:
stat, p_val = scipy.stats.ranksums(sample_x, sample_y, alternative='less')
statistics.append(stat)
p_vals.append(p_val)
d_score_statistics_loop_anchor = pd.DataFrame({
'classification_x': [name for ((name, _, _), _), _ in combos],
'chain_type_x': [name for ((_, name, _), _), _ in combos],
'cdr_x': [name for ((_, _, name), _), _ in combos],
'classification_y': [name for _, ((name, _, _), _) in combos],
'chain_type_y': [name for _, ((_, name, _), _) in combos],
'cdr_y': [name for _, ((_, _, name), _) in combos],
'statistic': statistics,
'p_val': p_vals,
'significant': [p_val < significance_level for p_val in p_vals],
})
d_score_statistics_loop_anchor['p_val'] = d_score_statistics_loop_anchor['p_val'].map(lambda num: f'{num:.2e}')
d_score_statistics_loop_anchor
0.008333333333333333
[27]:
| classification_x | chain_type_x | cdr_x | classification_y | chain_type_y | cdr_y | statistic | p_val | significant | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | anchor | alpha_chain | 1 | loop | alpha_chain | 1 | -1.220053 | 1.11e-01 | False |
| 1 | anchor | alpha_chain | 2 | loop | alpha_chain | 2 | -1.547078 | 6.09e-02 | False |
| 2 | anchor | alpha_chain | 3 | loop | alpha_chain | 3 | -5.328286 | 4.96e-08 | True |
| 3 | anchor | beta_chain | 1 | loop | beta_chain | 1 | 0.891960 | 8.14e-01 | False |
| 4 | anchor | beta_chain | 2 | loop | beta_chain | 2 | 1.596139 | 9.45e-01 | False |
| 5 | anchor | beta_chain | 3 | loop | beta_chain | 3 | -4.577162 | 2.36e-06 | True |
[28]:
combos = []
for pairing in list(itertools.combinations(treatments, 2)):
chain_type_x = pairing[0][0][1]
cdr_x = pairing[0][0][2]
chain_type_y = pairing[1][0][1]
cdr_y = pairing[1][0][2]
if (chain_type_x, cdr_x) == (chain_type_y, cdr_y):
combos.append(pairing)
significance_level = 0.05 / len(combos)
print(significance_level)
statistics = []
p_vals = []
for ((classification_x, chain_type_x, cdr_x), sample_x), ((classification_y, chain_type_y, cdr_y), sample_y) in combos:
stat, p_val = scipy.stats.ranksums(sample_x, sample_y, alternative='greater')
statistics.append(stat)
p_vals.append(p_val)
d_score_statistics_loop_anchor = pd.DataFrame({
'classification_x': [name for ((name, _, _), _), _ in combos],
'chain_type_x': [name for ((_, name, _), _), _ in combos],
'cdr_x': [name for ((_, _, name), _), _ in combos],
'classification_y': [name for _, ((name, _, _), _) in combos],
'chain_type_y': [name for _, ((_, name, _), _) in combos],
'cdr_y': [name for _, ((_, _, name), _) in combos],
'statistic': statistics,
'p_val': p_vals,
'significant': [p_val < significance_level for p_val in p_vals],
})
d_score_statistics_loop_anchor['p_val'] = d_score_statistics_loop_anchor['p_val'].map(lambda num: f'{num:.2e}')
d_score_statistics_loop_anchor
0.008333333333333333
[28]:
| classification_x | chain_type_x | cdr_x | classification_y | chain_type_y | cdr_y | statistic | p_val | significant | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | anchor | alpha_chain | 1 | loop | alpha_chain | 1 | -1.220053 | 8.89e-01 | False |
| 1 | anchor | alpha_chain | 2 | loop | alpha_chain | 2 | -1.547078 | 9.39e-01 | False |
| 2 | anchor | alpha_chain | 3 | loop | alpha_chain | 3 | -5.328286 | 1.00e+00 | False |
| 3 | anchor | beta_chain | 1 | loop | beta_chain | 1 | 0.891960 | 1.86e-01 | False |
| 4 | anchor | beta_chain | 2 | loop | beta_chain | 2 | 1.596139 | 5.52e-02 | False |
| 5 | anchor | beta_chain | 3 | loop | beta_chain | 3 | -4.577162 | 1.00e+00 | False |
Germline (CDR1 and CDR2) loops compared with CDR3 loops
[29]:
tcr_d_score_df['loop_type'] = tcr_d_score_df['cdr'].map(lambda cdr: 'Germline' if cdr in (1, 2) else 'CDR3')
[30]:
apo_holo_loop_type = (tcr_d_score_df.query("comparison == 'apo-holo'")
.groupby(['cdr_sequences_collated', 'loop_type', 'classification'],
dropna=False)['d_score']
.apply('sum')
.reset_index())
[31]:
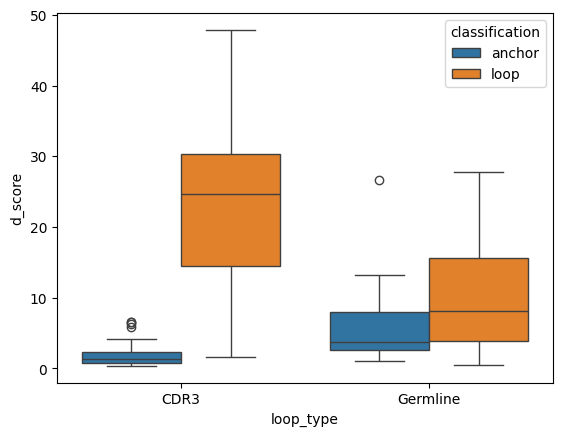
sns.boxplot(apo_holo_loop_type, x='loop_type', y='d_score', hue='classification')
[31]:
<AxesSubplot: xlabel='loop_type', ylabel='d_score'>
[32]:
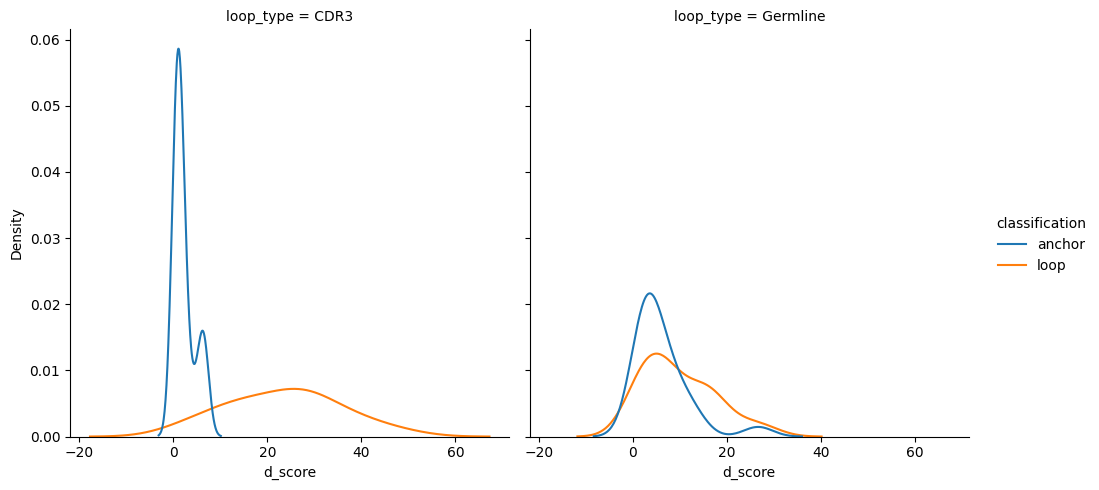
sns.displot(apo_holo_loop_type, x='d_score', col='loop_type', hue='classification', kind='kde')
[32]:
<seaborn.axisgrid.FacetGrid at 0x7fe3f2e5f280>
[33]:
treatment_options = ['classification', 'loop_type']
treatments = [(group, df['d_score'].to_numpy()) for group, df in apo_holo_loop_type.groupby(treatment_options)]
print(scipy.stats.kruskal(*[values for _, values in treatments]))
KruskalResult(statistic=46.51058594112732, pvalue=4.416965854082892e-10)
[34]:
combos = list(itertools.combinations(treatments, 2))
significance_level = 0.05 / len(combos)
print(significance_level)
statistics = []
p_vals = []
for ((classification_x, loop_type_x), sample_x), ((classification_y, loop_type_y), sample_y) in combos:
stat, p_val = scipy.stats.ranksums(sample_x, sample_y, alternative='two-sided')
statistics.append(stat)
p_vals.append(p_val)
d_score_statistics_loop_type = pd.DataFrame({
'classification_x': [name for ((name, _), _), _ in combos],
'loop_type_x': [name for ((_, name), _), _ in combos],
'classification_y': [name for _, ((name, _), _) in combos],
'loop_type_y': [name for _, ((_, name), _) in combos],
'statistic': statistics,
'p_val': p_vals,
'significant': [p_val < significance_level for p_val in p_vals],
})
d_score_statistics_loop_type['p_val'] = d_score_statistics_loop_type['p_val'].map(lambda num: f'{num:.2e}')
d_score_statistics_loop_type
0.008333333333333333
[34]:
| classification_x | loop_type_x | classification_y | loop_type_y | statistic | p_val | significant | |
|---|---|---|---|---|---|---|---|
| 0 | anchor | CDR3 | anchor | Germline | -3.591312 | 3.29e-04 | True |
| 1 | anchor | CDR3 | loop | CDR3 | -5.375231 | 7.65e-08 | True |
| 2 | anchor | CDR3 | loop | Germline | -4.037292 | 5.41e-05 | True |
| 3 | anchor | Germline | loop | CDR3 | -4.600635 | 4.21e-06 | True |
| 4 | anchor | Germline | loop | Germline | -1.713502 | 8.66e-02 | False |
| 5 | loop | CDR3 | loop | Germline | 3.708675 | 2.08e-04 | True |
Analysis of peptide D-scores
Load pMHC data
[35]:
pmhc_d_score_df = pd.read_csv(os.path.join(DATA_DIR, 'pmhc_per_res_apo_holo_d_score.csv'))
pmhc_d_score_df = pmhc_d_score_df.merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
).merge(
apo_holo_summary[['cdr_sequences_collated', 'peptide_sequence', 'mhc_slug']],
how='left',
left_on='complex_id',
right_index=True,
)
[36]:
d_score_df_holo = pd.read_csv(os.path.join(DATA_DIR, 'pmhc_per_res_holo_holo_d_score.csv'))
d_score_df_holo['mhc_slug'] = None
d_score_df_holo['peptide_sequence'] = None
mhc_pattern = r'^hla|h2'
mhc_alinged_complex_ids = d_score_df_holo['complex_id'].str.contains(mhc_pattern, regex=True)
mhc_slug_peptides = d_score_df_holo[mhc_alinged_complex_ids]['complex_id'].str.rsplit('_', n=1)
mhc_slugs = mhc_slug_peptides.map(lambda composite: composite[0])
peptides = mhc_slug_peptides.map(lambda composite: composite[1])
d_score_df_holo.loc[mhc_alinged_complex_ids, 'mhc_slug'] = mhc_slugs
d_score_df_holo.loc[mhc_alinged_complex_ids, 'peptide_sequence'] = peptides
d_score_df_holo = d_score_df_holo.merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_x_name',
right_on='file_name',
).merge(
apo_holo_summary[['file_name', 'pdb_id', 'structure_type', 'state']],
how='left',
left_on='structure_y_name',
right_on='file_name',
)
d_score_df_holo_pmhc = d_score_df_holo[mhc_alinged_complex_ids]
[37]:
pmhc_d_score_df = pd.concat([pmhc_d_score_df, d_score_df_holo_pmhc])
[38]:
pmhc_d_score_df['comparison'] = pmhc_d_score_df['state_x'] + '-' + pmhc_d_score_df['state_y']
pmhc_d_score_df['comparison'] = pmhc_d_score_df['comparison'].map(
lambda entry: 'apo-holo' if entry == 'holo-apo' else entry
)
[39]:
pmhc_d_score_df['structure_comparison'] = pmhc_d_score_df.apply(
lambda row: '-'.join(sorted([row.structure_x_name, row.structure_y_name])),
axis='columns',
)
pmhc_d_score_df = pmhc_d_score_df.drop_duplicates(['structure_comparison', 'chain_type', 'tcr_contact',
'residue_name', 'residue_seq_id', 'residue_insert_code'])
pmhc_d_score_df = pmhc_d_score_df.reset_index(drop=True)
[40]:
pmhc_d_score_df['peptide_positon'] = None
peptide_positions = (pmhc_d_score_df.query("chain_type == 'antigen_chain'")
.groupby(['complex_id', 'structure_x_name', 'structure_y_name'])
.cumcount() + 1)
pmhc_d_score_df.loc[pmhc_d_score_df['chain_type'] == 'antigen_chain', 'peptide_position'] = peptide_positions
[41]:
pmhc_d_score_df['peptide_length'] = pmhc_d_score_df['peptide_sequence'].str.len()
[42]:
pmhc_d_score_df = pmhc_d_score_df[~pmhc_d_score_df['d_score'].isnull()].reset_index(drop=True)
[43]:
pmhc_d_score_df = pmhc_d_score_df.groupby(['mhc_slug',
'peptide_sequence',
'comparison',
'chain_type',
'tcr_contact',
'residue_name',
'residue_seq_id',
'residue_insert_code',
'peptide_position',
'peptide_length'], dropna=False)['d_score'].apply('mean').reset_index()
[44]:
pmhc_d_score_df
[44]:
| mhc_slug | peptide_sequence | comparison | chain_type | tcr_contact | residue_name | residue_seq_id | residue_insert_code | peptide_position | peptide_length | d_score | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | h2_db | ASNENMETM | apo-apo | antigen_chain | False | ASN | 3 | NaN | 3.0 | 9 | 0.065791 |
| 1 | h2_db | ASNENMETM | apo-apo | antigen_chain | False | ASN | 5 | NaN | 5.0 | 9 | 0.174347 |
| 2 | h2_db | ASNENMETM | apo-apo | antigen_chain | False | GLU | 4 | NaN | 4.0 | 9 | 0.215112 |
| 3 | h2_db | ASNENMETM | apo-apo | antigen_chain | False | GLU | 7 | NaN | 7.0 | 9 | 0.015743 |
| 4 | h2_db | ASNENMETM | apo-apo | antigen_chain | False | MET | 6 | NaN | 6.0 | 9 | 0.019902 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 42111 | hla_e_01_03 | RLPAKAPLL | apo-holo | mhc_chain1 | True | THR | 1073 | NaN | NaN | 9 | 0.001763 |
| 42112 | hla_e_01_03 | RLPAKAPLL | apo-holo | mhc_chain1 | True | TRP | 1077 | NaN | NaN | 9 | 0.004126 |
| 42113 | hla_e_01_03 | RLPAKAPLL | apo-holo | mhc_chain1 | True | TYR | 59 | NaN | NaN | 9 | 0.015783 |
| 42114 | hla_e_01_03 | RLPAKAPLL | apo-holo | mhc_chain1 | True | TYR | 1070 | NaN | NaN | 9 | 0.004527 |
| 42115 | hla_e_01_03 | RLPAKAPLL | apo-holo | mhc_chain1 | True | VAL | 76 | NaN | NaN | 9 | 0.011465 |
42116 rows × 11 columns
Analysis
Comparing apo-apo, apo-holo, and holo-holo peptide D-scores
[45]:
peptide_d_scores = (pmhc_d_score_df.query("chain_type == 'antigen_chain'")
.groupby(['mhc_slug', 'peptide_sequence', 'comparison'], dropna=False)['d_score']
.apply('sum')
.reset_index())
[46]:
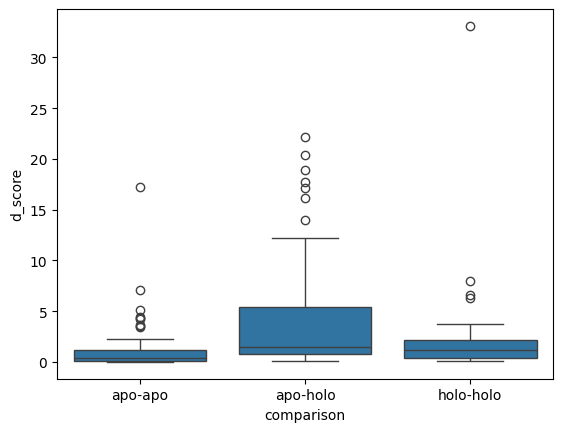
sns.boxplot(peptide_d_scores, x='comparison', y='d_score')
[46]:
<AxesSubplot: xlabel='comparison', ylabel='d_score'>
[47]:
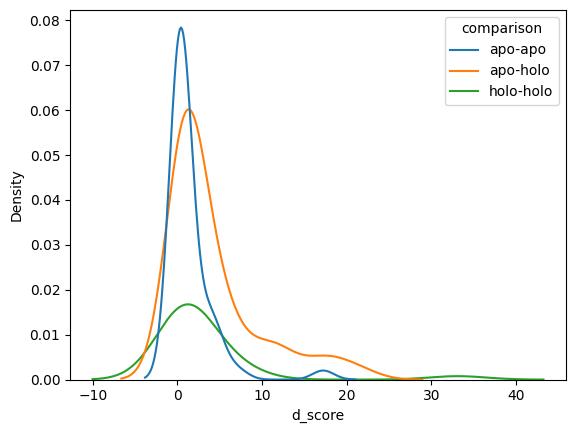
sns.kdeplot(peptide_d_scores, hue='comparison', x='d_score')
[47]:
<AxesSubplot: xlabel='d_score', ylabel='Density'>
[48]:
peptide_d_scores.groupby('comparison')['d_score'].std()
[48]:
comparison
apo-apo 2.788440
apo-holo 5.380116
holo-holo 6.488355
Name: d_score, dtype: float64
[49]:
treatments = [(group, df['d_score'].to_numpy()) for group, df in peptide_d_scores.groupby('comparison')]
print(scipy.stats.kruskal(*[values for _, values in treatments]))
KruskalResult(statistic=26.286830514084954, pvalue=1.958336328723338e-06)
[50]:
combos = list(itertools.combinations(treatments, 2))
significance_level = 0.05 / len(combos)
print(significance_level)
statistics = []
p_vals = []
for (comparison_x, sample_x), (comparison_y, sample_y) in combos:
stat, p_val = scipy.stats.ranksums(sample_x, sample_y, alternative='two-sided')
statistics.append(stat)
p_vals.append(p_val)
d_score_statistics_peptide = pd.DataFrame({
'comparison_x': [name for (name, _), _ in combos],
'comparison_y': [name for _, (name, _) in combos],
'statistic': statistics,
'p_val': p_vals,
'significant': [p_val < significance_level for p_val in p_vals],
})
d_score_statistics_peptide['p_val'] = d_score_statistics_peptide['p_val'].map(lambda num: f'{num:.2e}')
d_score_statistics_peptide
0.016666666666666666
[50]:
| comparison_x | comparison_y | statistic | p_val | significant | |
|---|---|---|---|---|---|
| 0 | apo-apo | apo-holo | -5.022685 | 5.10e-07 | True |
| 1 | apo-apo | holo-holo | -2.529214 | 1.14e-02 | True |
| 2 | apo-holo | holo-holo | 1.781758 | 7.48e-02 | False |
Per-residue (per length) D-scores of peptides
[51]:
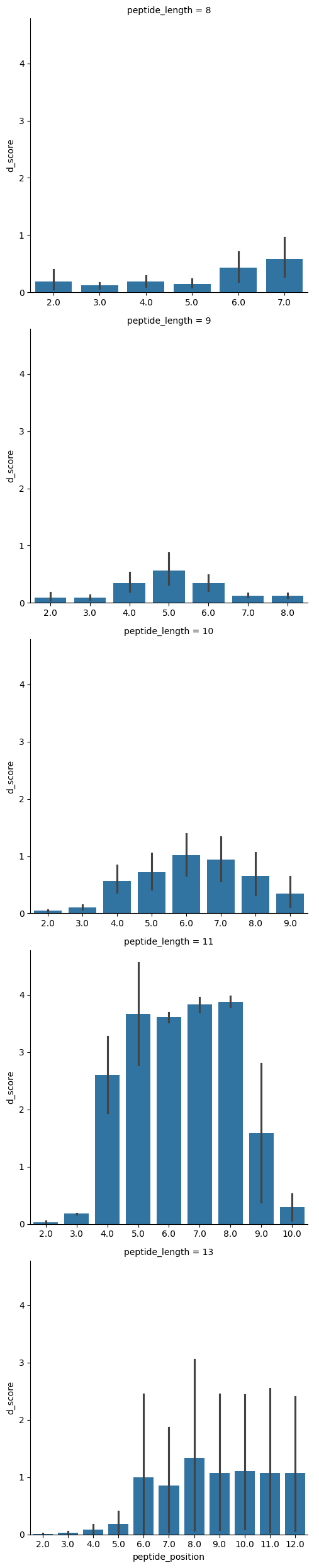
sns.catplot(pmhc_d_score_df.query("chain_type == 'antigen_chain'").sort_values('peptide_position'),
row='peptide_length',
x='peptide_position', y='d_score',
sharex=False,
kind='bar')
[51]:
<seaborn.axisgrid.FacetGrid at 0x7fe3f3400820>
Conclusion
Overall, the analysis of D-scores shows very similar results to the RMSD calculations done in other notebooks. The flexibility of TCRs seems apparent in the CDR3 loops, and the peptides have flexibility in the canonical TCR contact regions (p3 to pn-1).